
Development, Validation and Application of a Targeted LC-MS Method for Quantification of Microcystins and Nodularin: Towards a Better Characterization of Drinking Water

 , , and
, , and
Abstract
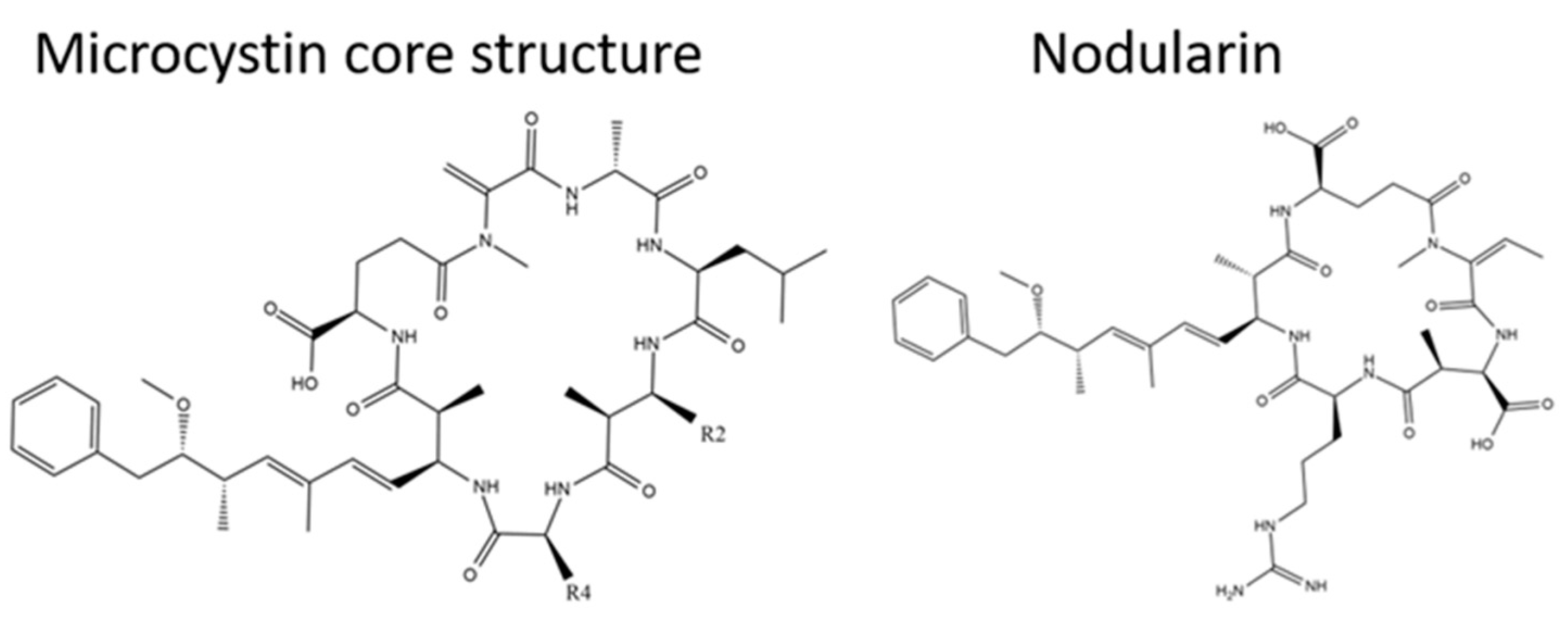
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Water Samples
2.3. Sample Preparation
2.4. UHPLC-MS/MS Conditions
2.5. Optimization of the MS/MS Conditions
2.6. Method Validation Procedure
3. Results
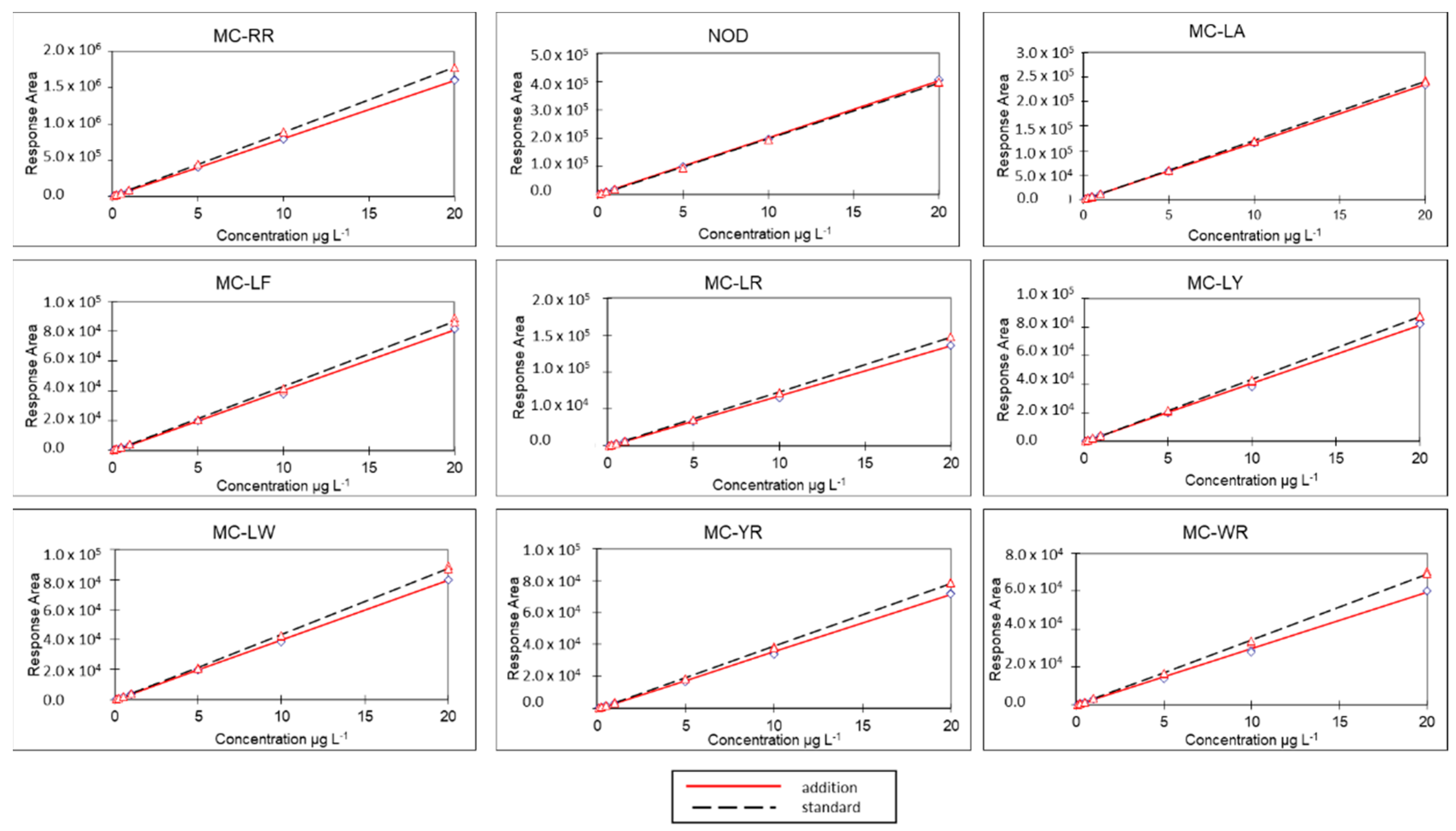
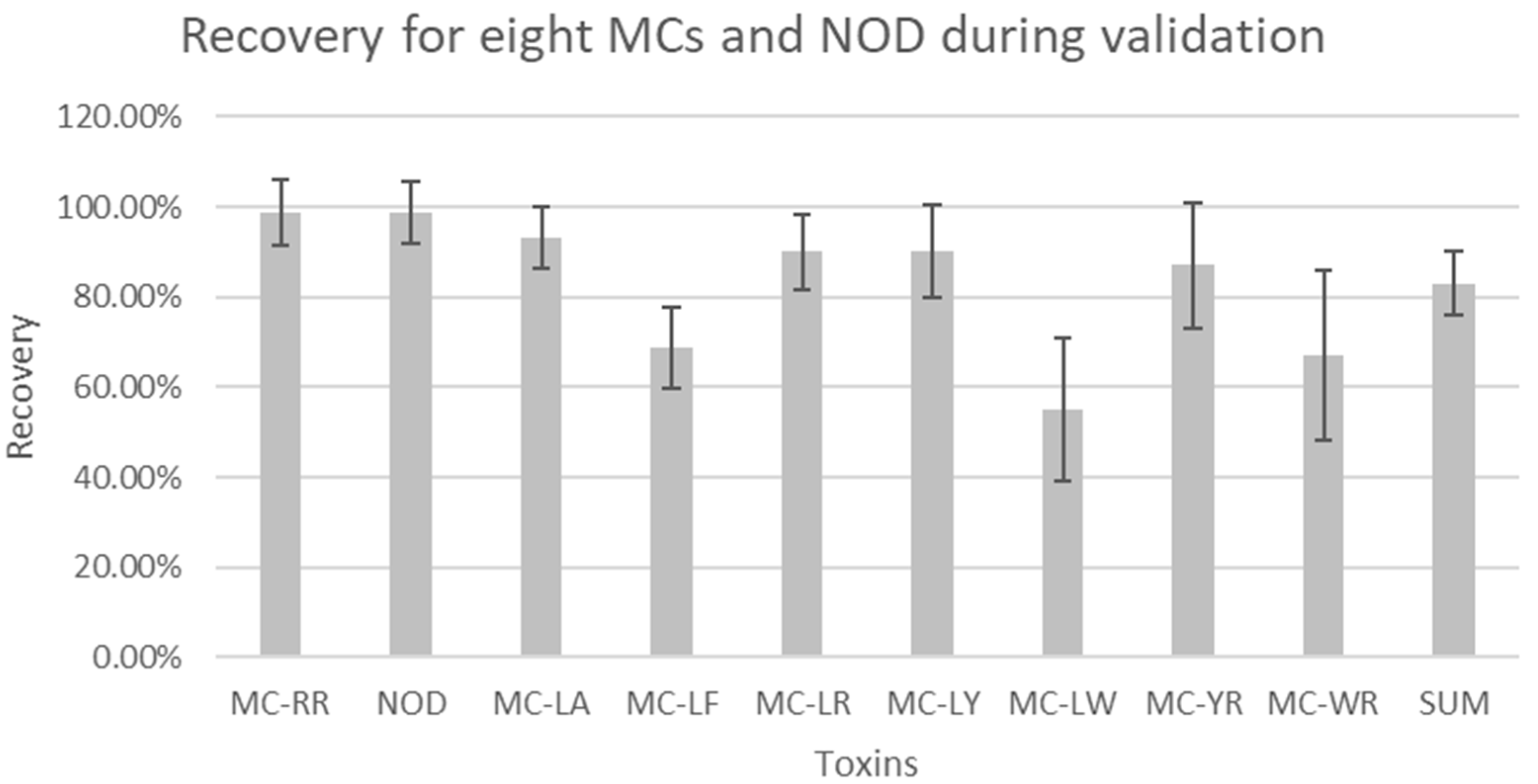
3.1. Method Validation
3.2. Application of the Method in Drinking Water
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boergens, E.; Güntner, A.; Dobslaw, H.; Dahle, C. Quantifying the Central European Droughts in 2018 and 2019 with GRACE Follow-On. Geophys. Res. Lett. 2020, 47, e2020GL087285. [Google Scholar] [CrossRef]
- Peeters, B. Waterverbruik Landbouw. 2021. Available online: https://www.vmm.be/sectoren/landbouw/waterverbruik-landbouw (accessed on 24 February 2022).
- Vlaamse Milieumaatschappij Drinkwaterbalans Voor Vlaanderen. 2018. Available online: https://www.vmm.be/publicaties/drinkwaterbalans-voor-vlaanderen-2018#:~:text=In%202018%20werd%20367%2C0,2018%205%25%20minder%20drinkwater%20geproduceerd (accessed on 24 February 2022).
- Vlaamse Milieumaatschappij Drinkwaterbalans Voor Vlaanderen. 2019. Available online: https://publicaties.vlaanderen.be/view-file/40084#:~:text=In%202019%20werd%20in%20totaal,m%C2%B3%20is%20grondwater%20uit%20Walloni%C3%AB (accessed on 16 November 2021).
- Direction des Eaux Souterraines. Direction de la Coordination des Données Qualite des Eaux Distribuees par le Reseau Public en Wallonie. 2018. Available online: http://environnement.wallonie.be/de/eso/eau_distribution/pdf/eau_distribution.pdf (accessed on 15 December 2020).
- Brahy, V.; Huart, M.; T’serstevens, J.-J. L’Exploitation des Ressources en Eau de Surface; Institut Numérique. 2013. Available online: https://www.institut-numerique.org/49-exploitation-des-ressources-en-eau-de-surface-52315bfe26de0 (accessed on 18 November 2021).
- État de l’Environnement Wallon. Production d’Eau de Distribution. Available online: http://etat.environnement.wallonie.be/contents/indicatorsheets/RESS%203.html (accessed on 18 November 2021).
- Peeters, B. Drinkwaterwinning. 2019. Available online: https://www.milieurapport.be/downloads/9156c60dfb7444158616fc887976dd97/1575968500/drinkwaterwinning.pdf (accessed on 15 December 2020).
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful Algal Blooms and Eutrophication: Nutrient Sources, Composition, and Consequences. Estuaries 2002, 25, 704–726. [Google Scholar] [CrossRef]
- Wagner, N.D.; Osburn, F.S.; Wang, J.; Taylor, R.B.; Boedecker, A.R.; Chambliss, C.K.; Brooks, B.W.; Scott, J.T. Biological Stoichiometry Regulates Toxin Production in Microcystis aeruginosa (UTEX 2385). Toxins 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descy, J.-P.; Leprieur, F.; Pirlot, S.; Leporcq, B.; Van Wichelen, J.; Peretyatko, A.; Teissier, S.; Codd, G.A.; Triest, L.; Vyverman, W.; et al. Identifying the Factors Determining Blooms of Cyanobacteria in a Set of Shallow Lakes. Ecol. Inform. 2016, 34, 129–138. [Google Scholar] [CrossRef]
- O’Neil, J.M.; Davis, T.W.; Burford, M.A.; Gobler, C.J. The Rise of Harmful Cyanobacteria Blooms: The Potential Roles of Eutrophication and Climate Change. Harmful Algae 2012, 14, 313–334. [Google Scholar] [CrossRef]
- Mantzouki, E.; Lürling, M.; Fastner, J.; de Senerpont Domis, L.; Wilk-Wo’zniak, E. Temperature Effects Explain Continental Scale Distribution of Cyanobacterial Toxins. Toxins 2018, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Mazur-Marzec, H.; Sutryk, K.; Kobos, J.; Hebel, A.; Hohlfeld, N.; Błaszczyk, A.; Toruńska, A.; Kaczkowska, M.J.; Łysiak-Pastuszak, E.; Kraśniewski, W.; et al. Occurrence of Cyanobacteria and Cyanotoxin in the Southern Baltic Proper. Filamentous Cyanobacteria versus Single-Celled Picocyanobacteria. Hydrobiologia 2013, 701, 235–252. [Google Scholar] [CrossRef] [Green Version]
- MacKintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial Microcystin-LR is a Potent and Specific Inhibitor of Protein Phosphatases 1 and 2A from Both Mammals and Higher Plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Runnegar, M.T.; Kong, S.; Berndt, N. Protein Phosphatase Inhibition and in Vivo Hepatotoxicity of Microcystins. Am. J. Physiol.-Gastrointest. Liver Physiol. 1993, 265, G224–G230. [Google Scholar] [CrossRef]
- Fischer, A.; Hoeger, S.J.; Stemmer, K.; Feurstein, D.J.; Knobeloch, D.; Nussler, A.; Dietrich, D.R. The Role of Organic Anion Transporting Polypeptides (OATPs/SLCOs) in the Toxicity of Different Microcystin Congeners in Vitro: A Comparison of Primary Human Hepatocytes and OATP-Transfected HEK293 Cells. Toxicol. Appl. Pharmacol. 2010, 245, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Dahlmann, J.; Rühl, A.; Hummert, C.; Liebezeit, G.; Carlsson, P.; Granéli, E. Different Methods for Toxin Analysis in the Cyanobacterium Nodularia Spumigena (Cyanophyceae). Toxicon 2001, 39, 1183–1190. [Google Scholar] [CrossRef]
- An, J.; Carmichael, W.W. Use of a Colorimetric Protein Phosphatase Inhibition Assay and Enzyme Linked Immunosorbent Assay for the Study of Microcystins and Nodularins. Toxicon 1994, 32, 1495–1507. [Google Scholar] [CrossRef]
- Svirčev, Z.; Lalić, D.; Bojadzija Savic, G.; Tokodi, N.; Drobac Backovic, D.; Chen, L.; Meriluoto, J.; Codd, G. Global Geographical and Historical Overview of Cyanotoxin Distribution and Cyanobacterial Poisonings. Arch. Toxicol. 2019, 93, 2429–2481. [Google Scholar] [CrossRef]
- World Health Organization. Cyanobacterial Toxins: Microcystins. Background Document for Development of WHO Guidelines for Drinking-Water Quality and Guidelines for Safe Recreational Water Environments; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The Toxicity of Cyanobacterial Toxins in the Mouse: I Microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cyanobacterial Toxins: Microcystin-LR in Drinking-Water WHO/SDE/WSH/03.04/57; WHO: Geneva, Switzerland, 1998. [Google Scholar]
- Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the Quality of Water Intended for Human Consumption; EUR-Lex: Brussels, Belgium, 2020.
- Lawton, L.A.; Edwards, C.; Codd, G.A. Extraction and High-Performance Liquid Chromatographic Method for the Determination of Microcystins in Raw and Treated Waters. Analyst 1994, 119, 1525. [Google Scholar] [CrossRef]
- Chu, F.S.; Huang, X.; Wei, R.D.; Carmichael, W.W. Production and Characterization of Antibodies against Microcystins. Appl. Environ. Microbiol. 1989, 55, 1928–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinteich, J.; Id, J.P.; Id, S.A.W.; Id, F.H.; Laughinghouse, H.D.; Id, I.V.; Pearce, D.A.; Dietrich, D.R.; Wilmotte, A. Toxic Cyanobacteria in Svalbard: Chemical Diversity of Microcystins Detected Using a Precursor Ion Screening Method. Toxins 2018, 10, 147. [Google Scholar] [CrossRef] [Green Version]
- Spoof, L.; Vesterkvist, P.; Lindholm, T.; Meriluoto, J. Screening for Cyanobacterial Hepatotoxins, Microcystins and Nodularin in Environmental Water Samples by Reversed-Phase Liquid Chromatography-Electrospray Ionisation Mass Spectrometry. J. Chromatogr. A 2003, 1020, 105–119. [Google Scholar] [CrossRef]
- Turner, A.D.; Waack, J.; Lewis, A.; Edwards, C.; Lawton, L. Development and Single-Laboratory Validation of a UHPLC-MS/MS Method for Quantitation of Microcystins and Nodularin in Natural Water, Cyanobacteria, Shellfish and Algal Supplement Tablet Powders. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1074–1075, 111–123. [Google Scholar] [CrossRef]
- Zervou, S.-K.; Christophoridis, C.; Kaloudis, T.; Triantis, T.M.; Hiskia, A. New SPE-LC-MS/MS Method for Simultaneous Determination of Multi-Class Cyanobacterial and Algal Toxins. J. Hazard. Mater. 2017, 323, 56–66. [Google Scholar] [CrossRef]
- Pekar, H.; Westerberg, E.; Bruno, O.; Lääne, A.; Persson, K.M.; Sundström, L.F.; Thim, A.M. Fast, Rugged and Sensitive Ultra High Pressure Liquid Chromatography Tandem Mass Spectrometry Method for Analysis of Cyanotoxins in Raw Water and Drinking Water—First Findings of Anatoxins, Cylindrospermopsins and Microcystin Variants in Swedish Source Wa. J. Chromatogr. A 2016, 1429, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.; Dammann, V.; Donnevert, G. Quality Assurance in Analytical Chemistry, 2nd ed.; WILEY: Weinheim, Germany, 2007. [Google Scholar]
- 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (Text with EEA Relevance) (Notified under Document Number C(2002) 3044). Available online: http://op.europa.eu/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en (accessed on 30 November 2020).
- Van Loco, J.; Elskens, M.; Croux, C.; Beernaert, H. Linearity of Calibration Curves: Use and Misuse of the Correlation Coefficient. Accredit. Qual. Assur. 2002, 7, 281–285. [Google Scholar] [CrossRef]
- Altaner, S.; Puddick, J.; Fessard, V.; Feurstein, D.; Zemskov, I.; Wittmann, V.; Dietrich, D.R. Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC-ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards. Toxins 2019, 11, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez-Quijada, L.; Guzmán-Guillén, R.; Prieto Ortega, A.I.; Llana-Ruíz-Cabello, M.; Campos, A.; Vasconcelos, V.; Jos, Á.; Cameán, A.M. New Method for Simultaneous Determination of Microcystins and Cylindrospermopsin in Vegetable Matrices by SPE-UPLC-MS/MS. Toxins 2018, 10, 406. [Google Scholar] [CrossRef] [Green Version]
- Altaner, S.; Puddick, J.; Wood, S.A.; Dietrich, D.R. Adsorption of Ten Microcystin Congeners to Common Laboratory-Ware is Solvent and Surface Dependent. Toxins 2017, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Vlaamse Milieu Maatschappij (VMM). Status Indicator Bevoorrading Leidingwater: Situatie op 24/09/2020. Available online: https://www.dewatergroep.be/nl-be/drinkwater/weetjes-en-tips/van-bron-tot-kraan/oppervlaktewater#:~:text=Van%20oppervlaktewater%20tot%20drinkwater&text=Die%20behandeling%20gebeurt%20in%20het,gebied%20en%20dus%20drinkbaar%20wordt (accessed on 24 February 2022).
- Dewatergroep Oppervlakte Water: Van Oppervlakte Water Naar Drink Water. Available online: https://www.dewatergroep.be/nl-be/drinkwater/weetjes-en-tips/van-bron-tot-kraan/oppervlaktewater (accessed on 24 February 2022).
- Pitois, F.; Fastner, J.; Pagotto, C.; Dechesne, M. Multi-Toxin Occurrences in Ten French Water Resource Reservoirs. Toxins 2018, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- USEPA. Recommendations for Public Water Systems to Manage Cyanotoxins in Drinking Water; USEPA: Washington, DC, USA, 2015. [Google Scholar]
- Hummert, C.; Reichelt, M.; Weiß, J.; Liebert, H.P.; Luckas, B. Identification of Microcystins in Cyanobacteria from the Bleiloch Former Drinking-Water Reservoir (Thuringia, Germany). Chemosphere 2001, 44, 1581–1588. [Google Scholar] [CrossRef]
- Teneva, I.; Mladenov, R.; Belkinova, D.; Dimitrova-Dyulgerova, I.; Dzhambazov, B. Phytoplankton Community of the Drinking Water Supply Reservoir Borovitsa (South Bulgaria) with an Emphasis on Cyanotoxins and Water Quality. Open Life Sci. 2010, 5, 231–239. [Google Scholar] [CrossRef]
- He, X.; Liu, Y.L.; Conklin, A.; Westrick, J.; Weavers, L.K.; Dionysiou, D.D.; Lenhart, J.J.; Mouser, P.J.; Szlag, D.; Walker, H.W. Toxic Cyanobacteria and Drinking Water: Impacts, Detection and Treatment. Harmful Algae 2016, 54, 174–193. [Google Scholar] [CrossRef]
- Beversdorf, L.J.; Rude, K.; Weirich, C.A.; Bartlett, S.L.; Seaman, M.; Kozik, C.; Biese, P.; Gosz, T.; Suha, M.; Stempa, C.; et al. Analysis of Cyanobacterial Metabolites in Surface and Raw Drinking Waters Reveals More than Microcystin. Water Res. 2018, 140, 280–290. [Google Scholar] [CrossRef]
- Czyżewska, W.; Piontek, M.; Łuszczyńska, K. The Occurrence of Potential Harmful Cyanobacteria and Cyanotoxins in the Obrzyca River (Poland), a Source of Drinking Water. Toxins 2020, 12, 284. [Google Scholar] [CrossRef] [PubMed]
- Ballot, A.; Swe, T.; Mjelde, M.; Cerasino, L.; Hostyeva, V.; Miles, C.O. Cylindrospermopsin- and Deoxycylindrospermopsin-Producing Raphidiopsis Raciborskii and Microcystin-Producing Microcystis spp. in Meiktila Lake, Myanmar. Toxins 2020, 12, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamele, I.J.; Vasconcelos, V. Microcystin Incidence in the Drinking Water of Mozambique: Challenges for Public Health Protection. Toxins 2020, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Ribau Teixeira, M.; Rosa, M.J. Neurotoxic and Hepatotoxic Cyanotoxins Removal by Nanofiltration. Water Res. 2006, 40, 2837–2846. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, S.J.; Shaw, G.; Hitzfeld, B.C.; Dietrich, D.R. Occurrence and Elimination of Cyanobacterial Toxins in Two Australian Drinking Water Treatment Plants. Toxicon 2004, 43, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, P.; Schumann, R.; Wong, S.H. Microcystin-LR Adsorption by Activated Carbon. J. Colloid Interface Sci. 2001, 240, 1–8. [Google Scholar] [CrossRef]
- Ho, L.; Lambling, P.; Bustamante, H.; Duker, P.; Newcombe, G. Application of Powdered Activated Carbon for the Adsorption of Cylindrospermopsin and Microcystin Toxins from Drinking Water Supplies. Water Res. 2011, 45, 2954–2964. [Google Scholar] [CrossRef]
- Huang, W.-J.; Cheng, B.-L.; Cheng, Y.-L. Adsorption of Microcystin-LR by Three Types of Activated Carbon. J. Hazard. Mater. 2007, 141, 115–122. [Google Scholar] [CrossRef]
- Cook, D.; Newcombe, G. Removal of Microcystin Variants with Powdered Activated Carbon. Water Supply 2002, 2, 201–207. [Google Scholar] [CrossRef]
- Cook, D.; Newcombe, G.; Sztajnbok, P. The Application of Powdered Activated Carbon for Mib and Geosmin Removal: Predicting Pac Doses in Four Raw Waters. Water Res. 2001, 35, 1325–1333. [Google Scholar] [CrossRef]
- Clasen, J.; Mischke, U.; Drikas, M.; Chow, C. An Improved Method for Detecting Electrophoretic Mobility of Algae during the Destabilisation Process of Flocculation: Flocculant Demand of Different Species and the Impact of DOC. J. Water Supply Res. Technol.-AQUA 2000, 49, 89–101. [Google Scholar] [CrossRef]
- Abbas, T.; Kajjumba, G.W.; Ejjada, M.; Masrura, S.U.; Marti, E.J.; Khan, E.; Jones-Lepp, T.L. Recent Advancements in the Removal of Cyanotoxins from Water Using Conventional and Modified Adsorbents—A Contemporary Review. Water 2020, 12, 2756. [Google Scholar] [CrossRef]
- Westrick, J.A.; Szlag, D.C.; Southwell, B.J.; Sinclair, J. A Review of Cyanobacteria and Cyanotoxins Removal/Inactivation in Drinking Water Treatment. Anal. Bioanal. Chem. 2010, 397, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Acero, J.L.; Rodriguez, E.; Meriluoto, J. Kinetics of Reactions between Chlorine and the Cyanobacterial Toxins Microcystins. Water Res. 2005, 39, 1628–1638. [Google Scholar] [CrossRef]
- Daly, R.I.; Ho, L.; Brookes, J.D. Effect of Chlorination on Microcystis Aeruginosa Cell Integrity and Subsequent Microcystin Release and Degradation. Environ. Sci. Technol. 2007, 41, 4447–4453. [Google Scholar] [CrossRef]
- Song, W.; Teshiba, T.; Rein, K.; O’Shea, K.E. Ultrasonically Induced Degradation and Detoxification of Microcystin-LR (Cyanobacterial Toxin). Environ. Sci. Technol. 2005, 39, 6300–6305. [Google Scholar] [CrossRef]
- Song, W.; de la Cruz, A.A.; Rein, K.; O’Shea, K.E. Ultrasonically Induced Degradation of Microcystin-LR and -RR: Identification of Products, Effect of PH, Formation and Destruction of Peroxides. Environ. Sci. Technol. 2006, 40, 3941–3946. [Google Scholar] [CrossRef] [Green Version]
- Hudder, A.; Song, W.; O’Shea, K.E.; Walsh, P.J. Toxicogenomic Evaluation of Microcystin-LR Treated with Ultrasonic Irradiation. Toxicol. Appl. Pharmacol. 2007, 220, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Wert, E.C.; Dong, M.M.; Rosario-Ortiz, F.L. Using Digital Flow Cytometry to Assess the Degradation of Three Cyanobacteria Species after Oxidation Processes. Water Res. 2013, 47, 3752–3761. [Google Scholar] [CrossRef]
- Newcombe, G.; Nicholson, B.C. Water Treatment Options for Dissolved Cyanotoxins. J. Water Supply Res. Technol.-AQUA 2004, 53, 227–239. [Google Scholar] [CrossRef]
- Newcombe, G.; Nicholson, B. Treatment Options for the Saxitoxin Class of Cyanotoxins. Water Supply 2002, 2, 271–275. [Google Scholar] [CrossRef]
- Sorlini, S.; Collivignarelli, C.; Carnevale Miino, M.; Caccamo, F.M.; Collivignarelli, M.C. Kinetics of Microcystin-LR Removal in a Real Lake Water by UV/H2O2 Treatment and Analysis of Specific Energy Consumption. Toxins 2020, 12, 810. [Google Scholar] [CrossRef] [PubMed]
- Coral, L.A.; Zamyadi, A.; Barbeau, B.; Bassetti, F.J.; Lapolli, F.R.; Prévost, M. Oxidation of Microcystis Aeruginosa and Anabaena Flos-Aquae by Ozone: Impacts on Cell Integrity and Chlorination by-Product Formation. Water Res. 2013, 47, 2983–2994. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Toxins | Precursor Ion | Quantifier Ion (m/z) | Collison Energy (eV) | Cone Voltage (V) | Qualifier Ion (m/z) | Collison Energy (eV) | Cone Voltage (V) |
|---|---|---|---|---|---|---|---|
| MC-LR | 995.4 | 135.0 | 70.0 | 80.0 | 213.1 | 60.0 | 80.0 |
| MC-RR | 519.8 | 134.8 | 30.0 | 50.0 | 107.2 | 60.0 | 50.0 |
| MC-YR | 1045.5 | 135.3 | 80.0 | 60.0 | 212.9 | 60.0 | 60.0 |
| MC-WR | 1068.4 | 135.3 | 70.0 | 100.0 | 213.1 | 60.0 | 100.0 |
| MC-LY | 1002.4 | 135.4 | 60.0 | 50.0 | 213.0 | 50.0 | 50.0 |
| MC-LA | 910.3 | 135.1 | 60.0 | 50.0 | 107.1 | 80.0 | 50.0 |
| MC-LF | 986.3 | 135.0 | 60.0 | 70.0 | 213.1 | 60.0 | 70.0 |
| MC-LW | 1025.4 | 134.9 | 60.0 | 60.0 | 213.1 | 50.0 | 60.0 |
| NOD | 825.2 | 134.9 | 50.0 | 80.0 | 102.7 | 90.0 | 80.0 |
| Toxins | Spiked Concentration (µg L−1) | Recovery (%) | Repeatability (%) | Reproducibility (%) | Measurement Uncertainty (%) | Average S/N for LOD (0.1 µg L−1) | Average S/N for LOQ (0.5 µg L−1) | R2 |
|---|---|---|---|---|---|---|---|---|
| MC-RR | 0.5 | 96.00 | 5.03 | 9.12 | 18.25 | 361.85 | 1691.95 | 1.00 |
| 2.5 | 97.00 | 2.04 | 4.99 | 9.98 | ||||
| 5.0 | 103.00 | 6.39 | 7.98 | 15.96 | ||||
| Average | 98.70 | 4.48 | 7.37 | 14.73 | ||||
| NOD | 0.5 | 95.00 | 5.23 | 7.14 | 14.29 | 193.38 | 7605.39 | 1.00 |
| 2.5 | 98.00 | 2.80 | 5.08 | 10.15 | ||||
| 5.0 | 103.00 | 7.26 | 8.10 | 16.20 | ||||
| Average | 98.70 | 5.10 | 6.77 | 13.55 | ||||
| MC-LA | 0.5 | 90.00 | 4.66 | 6.43 | 12.85 | 60.53 | 222.93 | 1.00 |
| 2.5 | 92.00 | 3.30 | 6.35 | 12.70 | ||||
| 5.0 | 97.00 | 7.21 | 7.84 | 15.67 | ||||
| Average | 93.00 | 5.06 | 6.87 | 13.74 | ||||
| MC-LF | 0.5 | 68.00 | 3.56 | 5.75 | 11.51 | 37.42 | 106.18 | 1.00 |
| 2.5 | 66.00 | 4.87 | 14.05 | 28.09 | ||||
| 5.0 | 72.00 | 7.04 | 7.04 | 14.08 | ||||
| Average | 68.70 | 5.15 | 8.95 | 17.89 | ||||
| MC-LR | 0.5 | 88.00 | 2.27 | 7.19 | 14.37 | 95.72 | 432.98 | 1.00 |
| 2.5 | 89.00 | 1.63 | 7.67 | 15.34 | ||||
| 5.0 | 93.00 | 7.35 | 10.07 | 20.14 | ||||
| Average | 90.00 | 3.75 | 8.31 | 16.62 | ||||
| MC-LY | 0.5 | 88.00 | 5.29 | 10.39 | 20.78 | 49.53 | 174.58 | 1.00 |
| 2.5 | 88.00 | 2.70 | 9.41 | 18.82 | ||||
| 5.0 | 94.00 | 6.11 | 10.94 | 21.88 | ||||
| Average | 90.00 | 4.70 | 10.25 | 20.49 | ||||
| MC-LW | 0.5 | 53.00 | 8.68 | 16.23 | 32.45 | 42.34 | 98.27 | 1.00 |
| 2.5 | 53.00 | 8.10 | 19.64 | 39.28 | ||||
| 5.0 | 59.00 | 6.01 | 11.16 | 22.32 | ||||
| Average | 55.00 | 7.60 | 15.67 | 31.35 | ||||
| MC-YR | 0.5 | 83.00 | 6.32 | 10.69 | 21.38 | 54.48 | 192.34 | 1.00 |
| 2.5 | 87.00 | 4.17 | 15.80 | 31.61 | ||||
| 5.0 | 91.00 | 5.30 | 15.34 | 30.67 | ||||
| Average | 87.00 | 5.26 | 13.94 | 27.89 | ||||
| MC-WR | 0.5 | 62.00 | 11.31 | 19.64 | 39.29 | 55.80 | 254.99 | 1.00 |
| 2.5 | 67.00 | 6.43 | 21.05 | 42.10 | ||||
| 5.0 | 72.00 | 3.59 | 16.14 | 32.28 | ||||
| Average | 67.00 | 7.11 | 18.94 | 37.89 | ||||
| SUM | 4.5 * | 80.00 | 4.32 | 5.27 | 10.54 | / | / | / |
| 22.5 * | 82.00 | 2.99 | 8.59 | 17.18 | ||||
| 45.0 * | 87.00 | 5.92 | 7.08 | 14.15 | ||||
| Average | 83.00 | 4.41 | 6.98 | 13.96 |
| MC-RR | NOD | MC-LA | MC-LF | MC-LR | MC-LY | MC-LW | MC-YR | MC-WR | |
|---|---|---|---|---|---|---|---|---|---|
| t (b) | 53.59 | 3.34 | 7.92 | 5.57 | 11.54 | 7.53 | 11.93 | 9.17 | 12.02 |
| t (95%) | 2.06 | 2.06 | 2.06 | 2.06 | 2.06 | 2.06 | 2.06 | 2.06 | 2.06 |
| MC-RR | NOD | MC-LA | MC-LF | MC-LR | MC-LY | MC-LW | MC-YR | MC-WR |
|---|---|---|---|---|---|---|---|---|
| 97.00% | 90.00% | 81.00% | 61.00% | 72.00% | 74.00% | 43.00% | 77.00% | 60.00% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Hassel, W.H.R.; Huybrechts, B.; Masquelier, J.; Wilmotte, A.; Andjelkovic, M. Development, Validation and Application of a Targeted LC-MS Method for Quantification of Microcystins and Nodularin: Towards a Better Characterization of Drinking Water. Water 2022, 14, 1195. https://doi.org/10.3390/w14081195
Van Hassel WHR, Huybrechts B, Masquelier J, Wilmotte A, Andjelkovic M. Development, Validation and Application of a Targeted LC-MS Method for Quantification of Microcystins and Nodularin: Towards a Better Characterization of Drinking Water. Water. 2022; 14(8):1195. https://doi.org/10.3390/w14081195
Chicago/Turabian StyleVan Hassel, Wannes Hugo R., Bart Huybrechts, Julien Masquelier, Annick Wilmotte, and Mirjana Andjelkovic. 2022. "Development, Validation and Application of a Targeted LC-MS Method for Quantification of Microcystins and Nodularin: Towards a Better Characterization of Drinking Water" Water 14, no. 8: 1195. https://doi.org/10.3390/w14081195