
Microbial Community Structure and Bacterial Lineages Associated with Sulfonamides Resistance in Anthropogenic Impacted Larut River

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
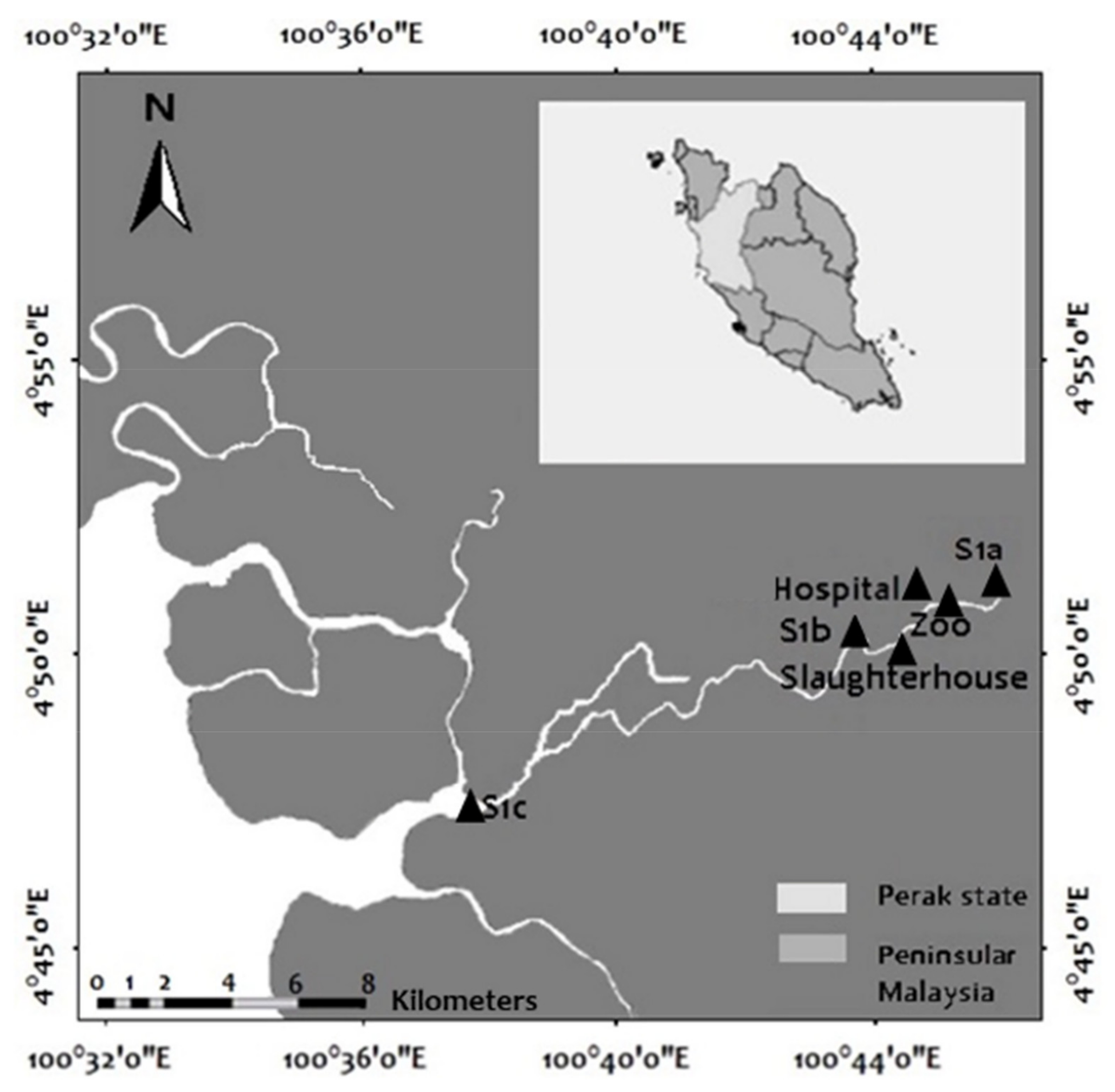
2.1. Sampling Sites
2.2. Metagenomics DNA Extraction
2.3. Microbial Community Structure
2.4. Identification of Culturable Sulfonamides-Resistant Bacteria
2.5. Detection of SAR Genes (sul1, sul2 and sul3) in the Sulfonamides-Resistant Bacteria
2.6. Statistical Analyses
3. Results and Discussion
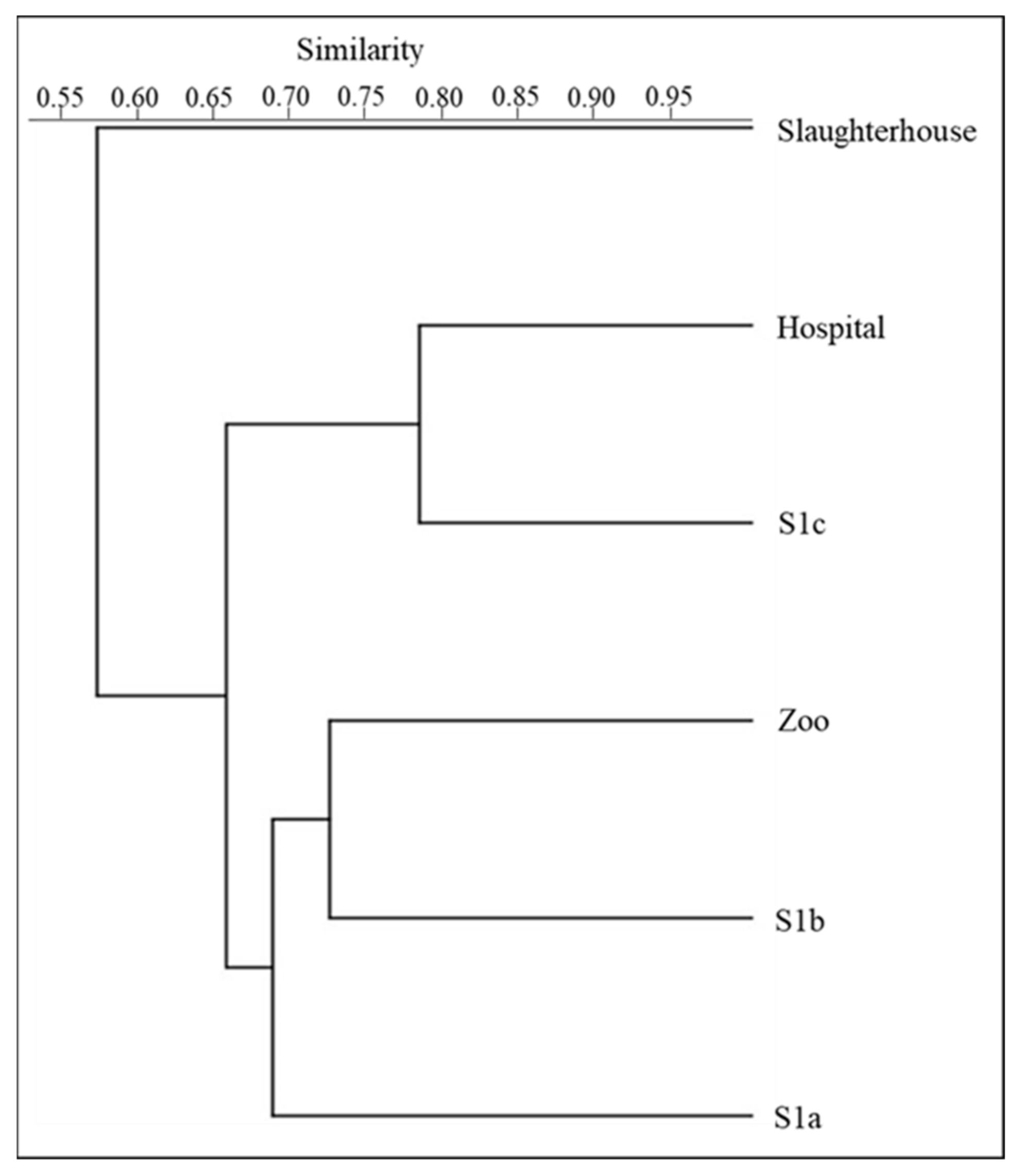
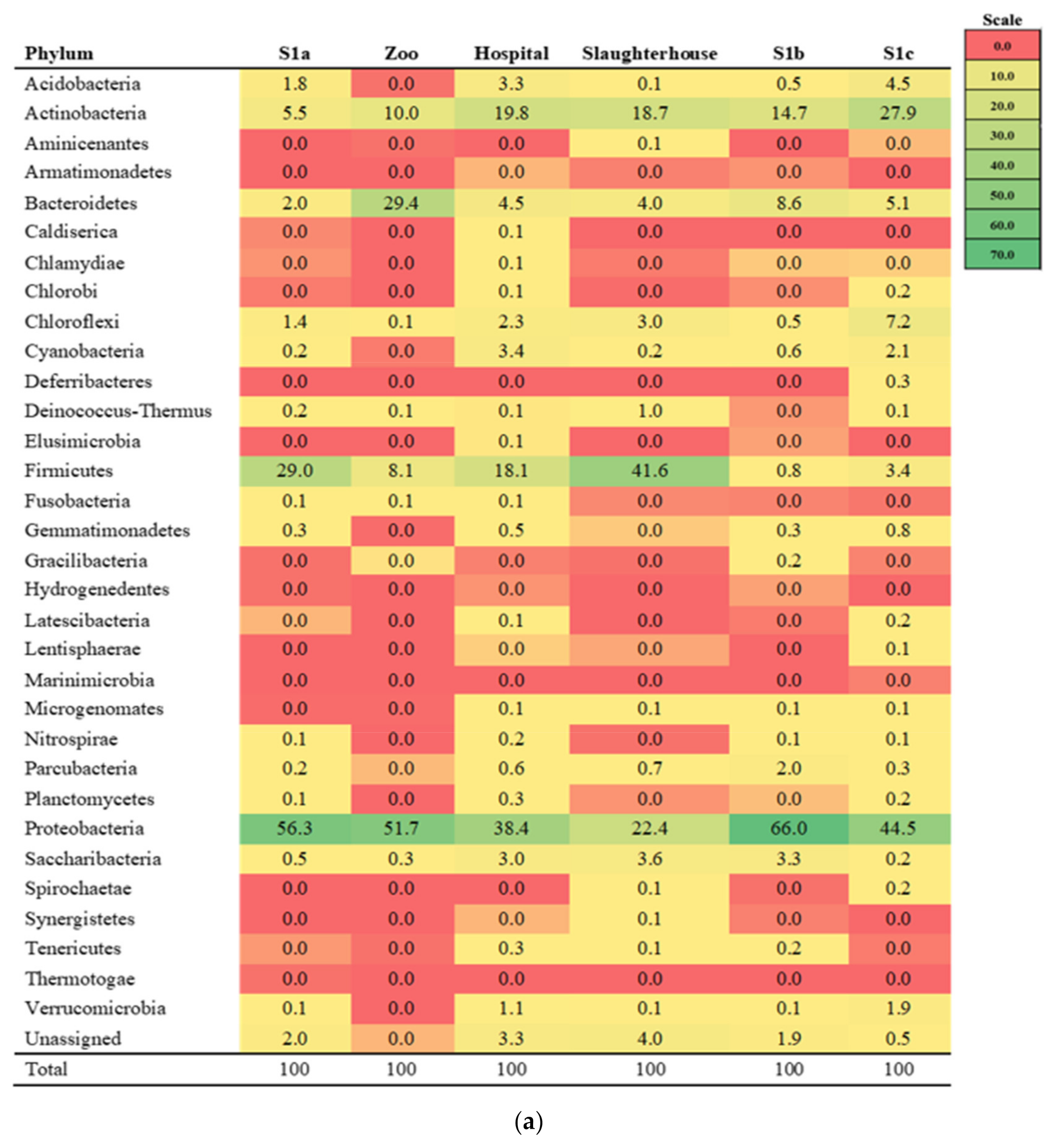
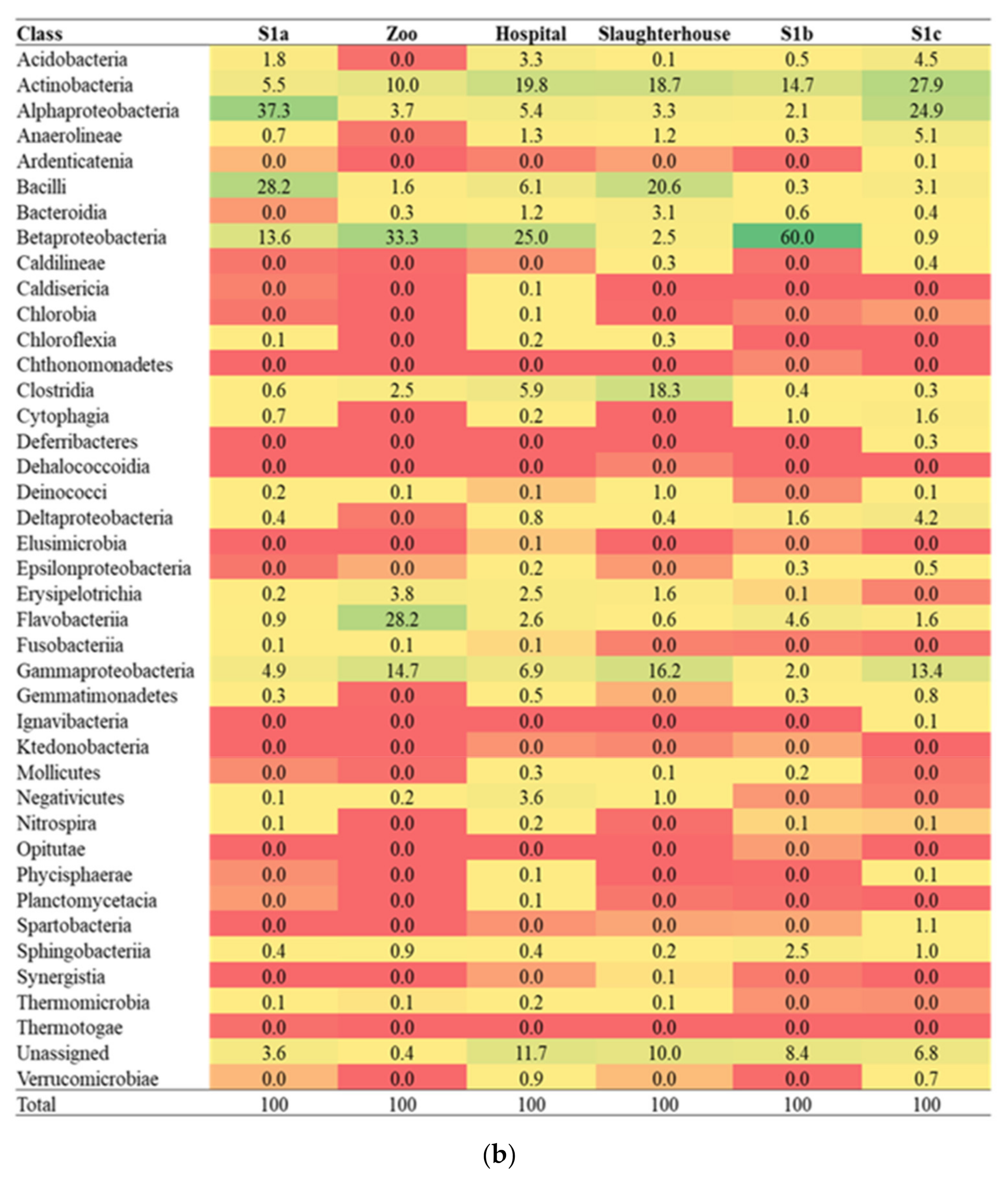
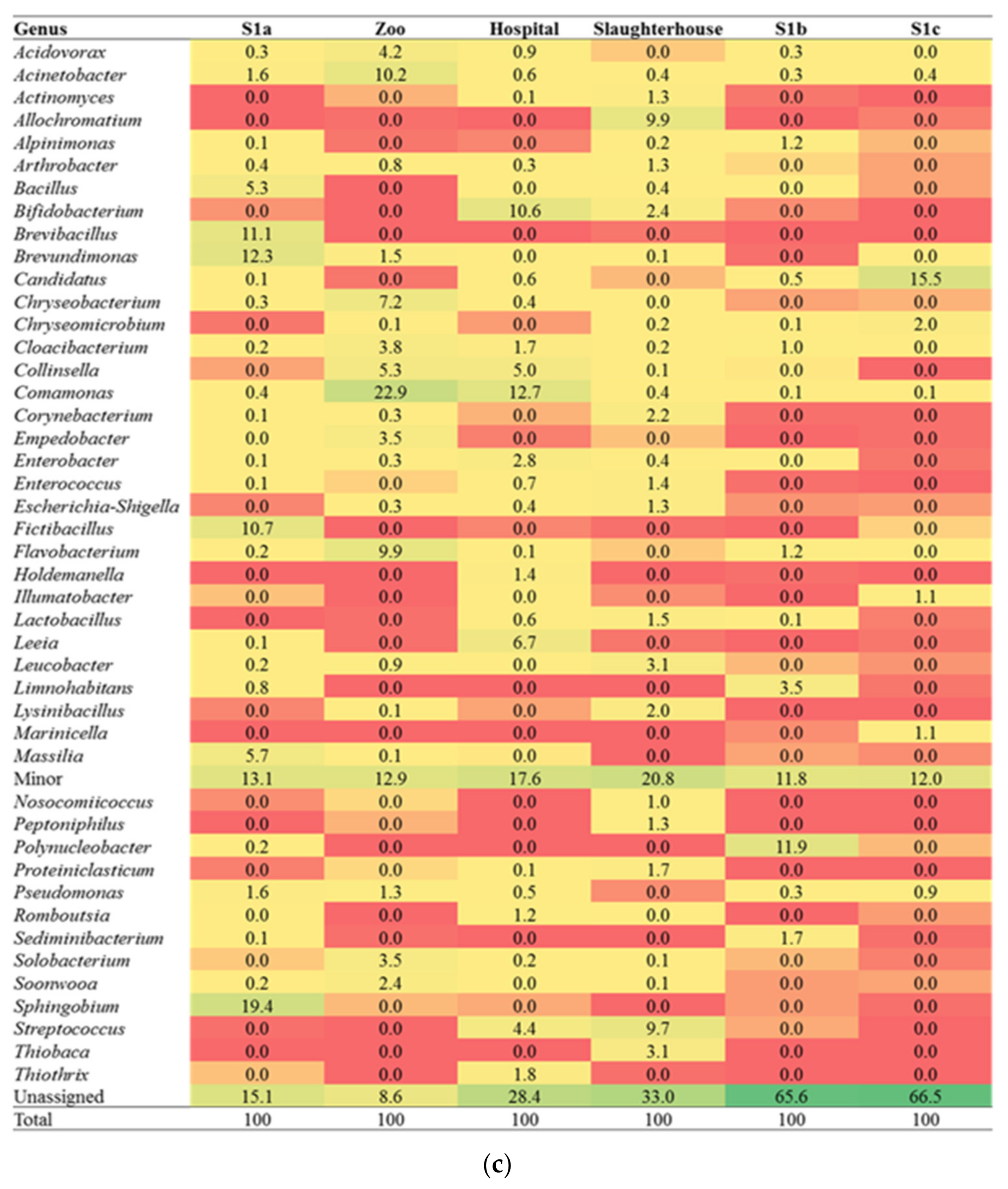
3.1. Diversity of the Microbial Community
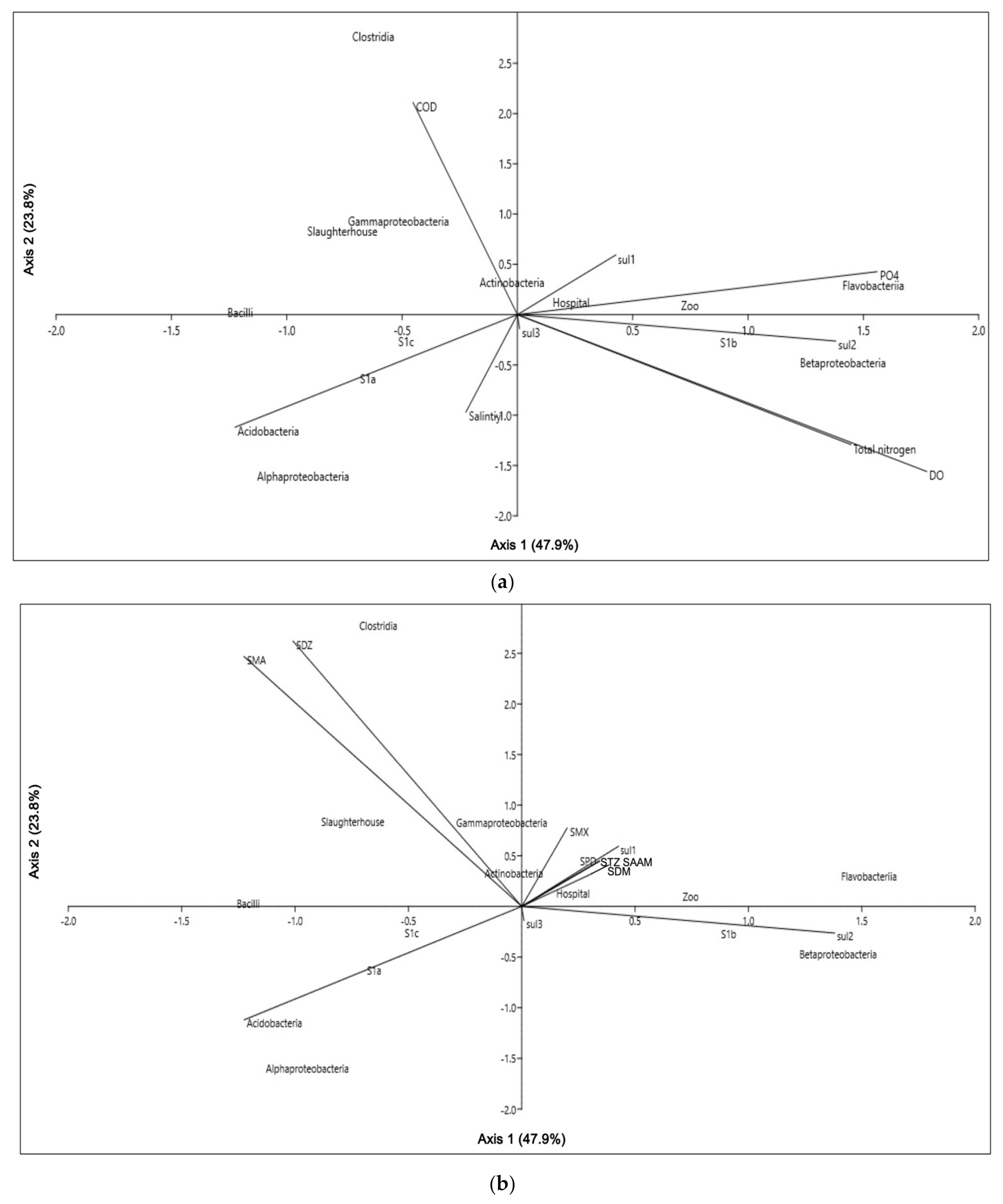
3.2. Environmental Physicochemical Parameters, Sulfonamides Residues and Their Relations to Microbial Community
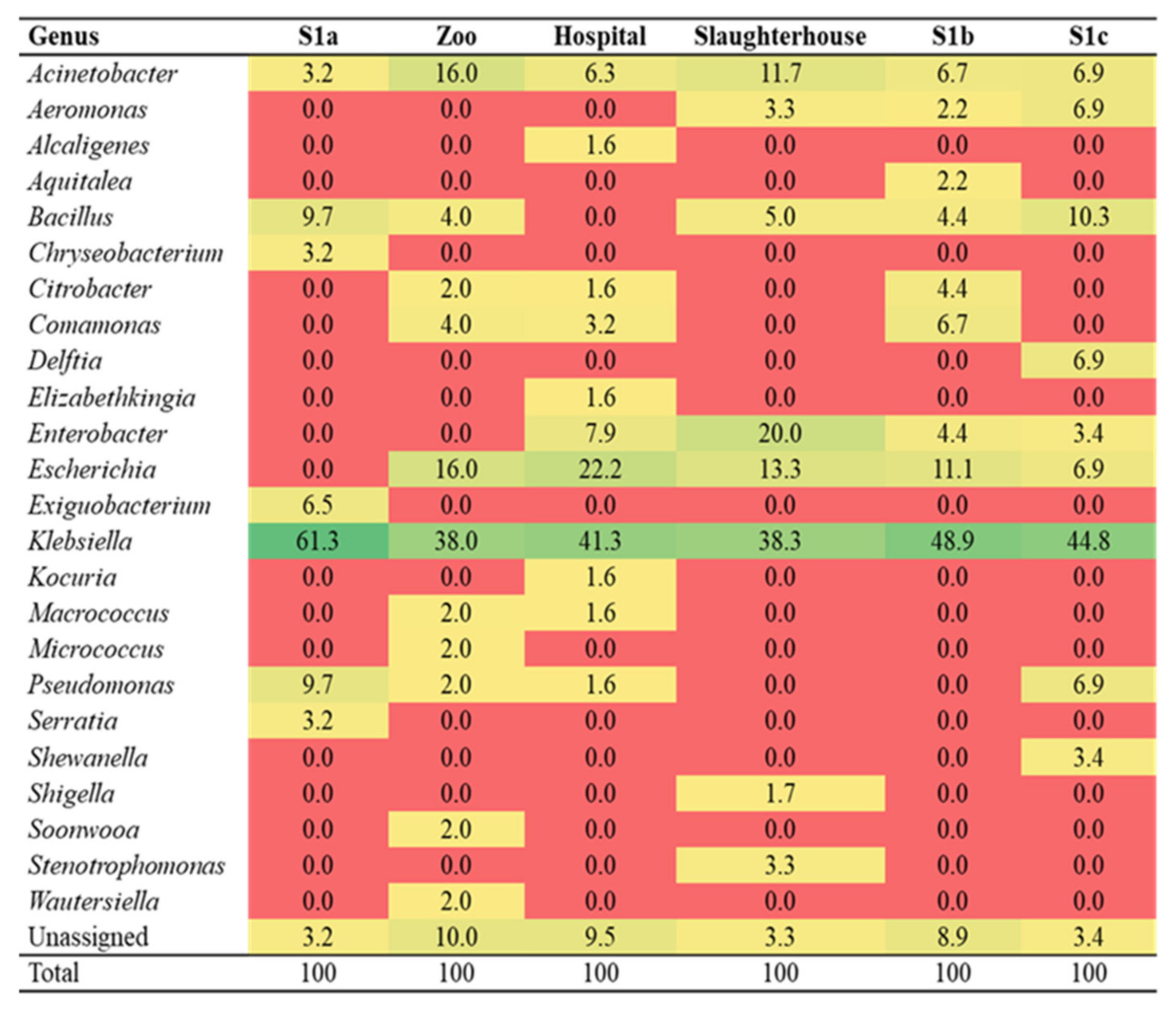
3.3. Identification of Culturable Sulfonamide-Resistant Bacteria
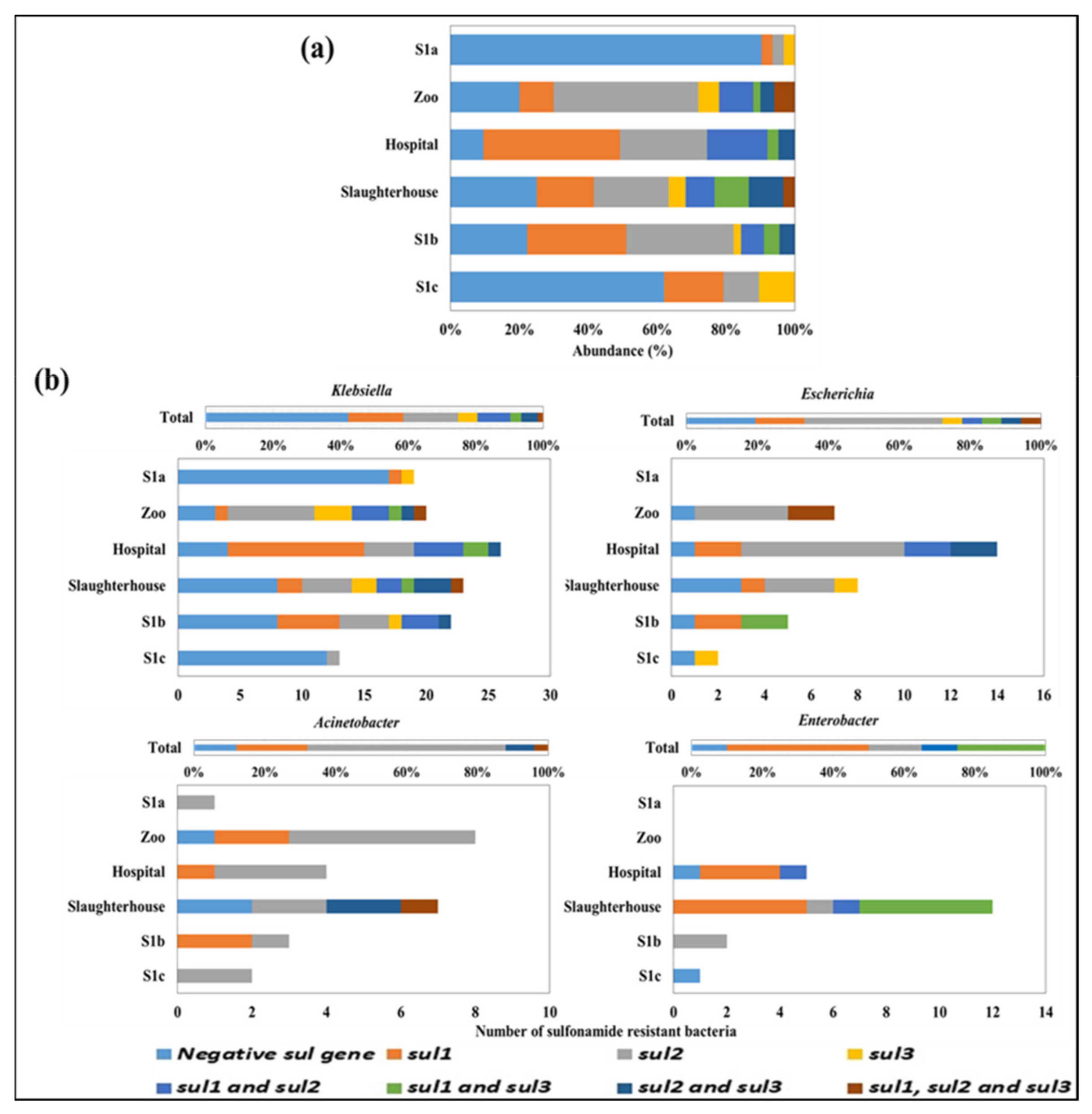
3.4. Detection of sul1, sul2, and sul3 Genes in the Culturable Sulfonamide-Resistant Isolates
3.5. Identification of Potential sul Genes Bacterial Carrier
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.A.; Dantas, G. The Shared Antibiotic Resistome of Soil Bacteria and Human Pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, M.K.; Forsberg, K.; Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2014, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.T.; Giovannoni, S.J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996, 62, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Qi, R.; Yang, M.; Zhang, Y.; Yu, T. Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 2011, 45, 6063–6073. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martinez, J.L.; Cantón, R. Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Rodriguez-Martinez, J.-M.; Mammeri, H.; Liard, A.; Nordmann, P. Origin of Plasmid-Mediated Quinolone Resistance Determinant QnrA. Antimicrob. Agents Chemother. 2005, 49, 3523–3525. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, L.; Manaia, C.; Merlin, C.; Schwartz, T.; Dagot, C.; Ploy, M.C.; Michael, I.; Fatta-Kassinos, D. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: A review. Sci. Total Environ. 2013, 447, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Handel, A.; Regoes, R.R.; Antia, R. The Role of Compensatory Mutations in the Emergence of Drug Resistance. PLoS Comput. Biol. 2006, 2, e137. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, I.R.; Watanabe, N.; Harter, T.; Glaser, B.; Radke, M. Effect of sulfonamide antibiotics on microbial diversity and activity in a Californian Mollic Haploxeralf. J. Soils Sediments 2010, 10, 537–544. [Google Scholar] [CrossRef]
- Economou, V.; Gousia, P. Agriculture and food animals as a source of antimicrobial-resistant bacteria. Infect. Drug Resist. 2015, 8, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Laffite, A.; Kilunga, P.I.; Kayembe, J.M.; Devarajan, N.; Mulaji, C.; Giuliani, G.; Slaveykova, V.; Poté, J. Hospital Effluents Are One of Several Sources of Metal, Antibiotic Resistance Genes, and Bacterial Markers Disseminated in Sub-Saharan Urban Rivers. Front. Microbiol. 2016, 7, 1128. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, K.M.; Wassermann, T.; Jensen, P.Ø.; Hengzuang, W.; Molin, S.; Høiby, N.; Ciofu, O. Sublethal Ciprofloxacin Treatment Leads to Rapid Development of High-Level Ciprofloxacin Resistance during Long-Term Experimental Evolution of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4215–4221. [Google Scholar] [CrossRef] [Green Version]
- Lye, Y.L.; Bong, C.W.; Lee, C.W.; Zhang, R.J.; Zhang, G.; Suzuki, S.; Chai, L.C. Anthropogenic impacts on sulfonamide residues and sulfonamide resistant bacteria and genes in Larut and Sangga Besar River, Perak. Sci. Total Environ. 2019, 688, 1335–1347. [Google Scholar] [CrossRef]
- Bien, T.L.T.; Sato-Takabe, Y.; Ogo, M.; Usui, M.; Suzuki, S. Persistence of Multi-Drug Resistance Plasmids in Sterile Water under Very Low Concentrations of Tetracycline. Microbes Environ. 2015, 30, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Baena, A.M.; Caicedo-Bejarano, L.D.; Chávez-Vivas, M. Structure of Bacterial Community with Resistance to Anti-biotics in Aquatic Environments. A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 2348. [Google Scholar] [CrossRef]
- Girijan, S.K.; Paul, R.; Rejish Kumar, V.J.; Pillai, D. Investigating the impact of hospital antibiotic usage on aquatic environment and aquaculture systems: A molecular study of quinolone resistance in Escherichia coli. Sci. Total Environ. 2020, 748, 141538. [Google Scholar] [CrossRef]
- Narciso-Da-Rocha, C.; Rocha, J.; Vaz-Moreira, I.; Lira, F.; Tamames, J.; Henriques, I.; Martinez, J.L.; Manaia, C.M. Bacterial lineages putatively associated with the dissemination of antibiotic resistance genes in a full-scale urban wastewater treatment plant. Environ. Int. 2018, 118, 179–188. [Google Scholar] [CrossRef]
- Sarmah, A.K.; Meyer, M.; Boxall, A.B. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 2006, 65, 725–759. [Google Scholar] [CrossRef]
- Lamshöft, M.; Sukul, P.; Zühlke, S.; Spiteller, M. Metabolism of 14C-labelled and non-labelled sulfadiazine after administration to pigs. Anal. Bioanal. Chem. 2007, 388, 1733–1745. [Google Scholar] [CrossRef]
- Manzetti, S.; Ghisi, R. The environmental release and fate of antibiotics. Mar. Pollut. Bull. 2014, 79, 7–15. [Google Scholar] [CrossRef]
- Sköld, O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Esuzuki, S.; Eogo, M.; Miller, T.W.; Eshimizu, A.; Etakada, H.; Siringan, M.A.T. Who possesses drug resistance genes in the aquatic environment? Sulfamethoxazole (SMX) resistance genes among the bacterial community in water environment of Metro-Manila, Philippines. Front. Microbiol. 2013, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef] [Green Version]
- Islas-Espinoza, M.; Reid, B.J.; Wexler, M.; Bond, P.L. Soil Bacterial Consortia and Previous Exposure Enhance the Biodegradation of Sulfonamides from Pig Manure. Microb. Ecol. 2012, 64, 140–151. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the Effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-Based Studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Wang, L.; Zhang, J.; Li, H.; Yang, H.; Peng, C.; Peng, Z.; Lu, L. Shift in the microbial community composition of surface water and sediment along an urban river. Sci. Total Environ. 2018, 627, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Na, G.; Gao, H.; Wang, L.; Bao, C.; Yao, Z. Fate of sulfonamide resistance genes in estuary environment and effect of anthropogenic activities. Sci. Total Environ. 2015, 527–528, 429–438. [Google Scholar] [CrossRef]
- Hoa, P.T.P.; Nonaka, L.; Viet, P.H.; Suzuki, S. Detection of the sul1, sul2, and sul3 genes in sulfonamide-resistant bacteria from wastewater and shrimp ponds of north Vietnam. Sci. Total Environ. 2008, 405, 377–384. [Google Scholar] [CrossRef]
- Kerrn, M.B.; Klemmensen, T.; Frimodt-Møller, N.; Espersen, F. Susceptibility of Danish Escherichia coli strains isolated from urinary tract infections and bacteraemia, and distribution of sul genes conferring sulphonamide resistance. J. Antimicrob. Chemother. 2002, 50, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Na, G.; Zhang, W.; Zhou, S.; Gao, H.; Lu, Z.; Wu, X.; Li, R.; Qiu, L.; Cai, Y.; Yao, Z. Sulfonamide antibiotics in the Northern Yellow Sea are related to resistant bacteria: Implications for antibiotic resistance genes. Mar. Pollut. Bull. 2014, 84, 70–75. [Google Scholar] [CrossRef]
- Dianawati, R.I.; Wahyuningsih, N.E.; Nur, M. Treatment of hospital waste water by ozone technology. J. Phys. Conf. Ser. 2018, 1025, 012013. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.; Vieira, R.P.; Cardoso, A.M.; Paranhos, R.; Albano, R.; Martins, O.B. Influence of Salinity on Bacterioplankton Communities from the Brazilian Rain Forest to the Coastal Atlantic Ocean. PLoS ONE 2011, 6, e17789. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, D.; Zhang, Y.; Shen, J.; van der Gast, C.; Hahn, M.W.; Wu, Q. Do Patterns of Bacterial Diversity along Salinity Gradients Differ from Those Observed for Macroorganisms? PLoS ONE 2011, 6, e27597. [Google Scholar] [CrossRef] [Green Version]
- Aldunate, M.; De la Iglesia, R.; Bertagnolli, A.D.; Ulloa, O. Oxygen modulates bacterial community composition in the coastal upwelling waters off central Chile. Deep Sea Res. Part II Top. Stud. Oceanogr. 2018, 156, 68–79. [Google Scholar] [CrossRef]
- Spietz, R.; Williams, C.M.; Rocap, G.; Horner-Devine, C. A Dissolved Oxygen Threshold for Shifts in Bacterial Community Structure in a Seasonally Hypoxic Estuary. PLoS ONE 2015, 10, e0135731. [Google Scholar] [CrossRef] [Green Version]
- Szekeres, E.; Baricz, A.; Chiriac, C.M.; Farkas, A.; Opris, O.; Soran, M.-L.; Andrei, A.-S.; Rudi, K.; Balcázar, J.L.; Dragos, N.; et al. Abundance of antibiotics, antibiotic resistance genes and bacterial community composition in wastewater effluents from different Romanian hospitals. Environ. Pollut. 2017, 225, 304–315. [Google Scholar] [CrossRef]
- Vaz-Moreira, I.; Nunes, O.; Manaia, C.M. Bacterial diversity and antibiotic resistance in water habitats: Searching the links with the human microbiome. FEMS Microbiol. Rev. 2014, 38, 761–778. [Google Scholar] [CrossRef]
- Wang, X.; Gu, J.; Gao, H.; Qian, X.; Li, H. Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China. Int. J. Environ. Res. Public Health 2018, 15, 708. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Zhenling, Z.; Wang, M.; Zeng, Z. Antibiotics, Antibiotic Resistance Genes, and Bacterial Community Composition in Fresh Water Aquaculture Environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef]
- Xu, K.; Wang, J.; Gong, H.; Li, Y.; Zhou, L.; Yan, M. Occurrence of antibiotics and their associations with antibiotic resistance genes and bacterial communities in Guangdong coastal areas. Ecotoxicol. Environ. Saf. 2019, 186, 109796. [Google Scholar] [CrossRef]
- Bondarczuk, K.; Piotrowska-Seget, Z. Microbial diversity and antibiotic resistance in a final effluent-receiving lake. Sci. Total Environ. 2018, 650, 2951–2961. [Google Scholar] [CrossRef]
- Drury, B.; Rosi-Marshall, E.; Kelly, J.J. Wastewater Treatment Effluent Reduces the Abundance and Diversity of Benthic Bacterial Communities in Urban and Suburban Rivers. Appl. Environ. Microbiol. 2013, 79, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Khandeparker, L.; Kuchi, N.; Kale, D.; Anil, A.C. Microbial community structure of surface sediments from a tropical estuarine environment using next generation sequencing. Ecol. Indic. 2016, 74, 172–181. [Google Scholar] [CrossRef]
- Narayan, A.; Patel, V.; Singh, P.; Patel, A.; Jain, K.; Karthikeyan, K.; Shah, A.; Madamwar, D. Response of microbial community structure to seasonal fluctuation on soils of Rann of Kachchh, Gujarat, India: Representing microbial dynamics and functional potential. Ecol. Genet. Genom. 2018, 6, 22–32. [Google Scholar] [CrossRef]
- Qiu, W.; Sun, J.; Fang, M.; Luo, S.; Tian, Y.; Dong, P.; Xu, B.; Zheng, C. Occurrence of antibiotics in the main rivers of Shenzhen, China: Association with antibiotic resistance genes and microbial community. Sci. Total Environ. 2019, 653, 334–341. [Google Scholar] [CrossRef]
- Szekeres, E.; Chiriac, C.; Baricz, A.; Szőke-Nagy, T.; Lung, I.; Soran, M.-L.; Rudi, K.; Dragos, N.; Coman, C. Investigating antibiotics, antibiotic resistance genes, and microbial contaminants in groundwater in relation to the proximity of urban areas. Environ. Pollut. 2018, 236, 734–744. [Google Scholar] [CrossRef]
- Tiquia, S.; Schleibak, M.; Schlaff, J.; Floyd, C.; Benipal, B.; Zakhem, E.; Murray, K. Microbial Community Profiling and Characterization of Some Heterotrophic Bacterial Isolates from River Waters and Shallow Groundwater Wells Along the Rouge River, Southeast Michigan. Environ. Technol. 2008, 29, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Jaguś, A. Assessment of the effectiveness of tresna reservoir protection based on the soła river waters contamination. Econ. Eng. 2017, 18, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Allgaier, M.; Brückner, S.; Jaspers, E.; Grossart, H.-P. Intra- and inter-lake variability of free-living and particle-associated Actinobacteria communities. Environ. Microbiol. 2007, 9, 2728–2741. [Google Scholar] [CrossRef] [PubMed]
- Kurtböke, D.I. Ecology and Habitat Distribution of Actinobacteria. In Biology and Biotechnology of Actinobacteria; Wink, J., Mohammadipanah, F., Hamedi, J., Eds.; Springer: Cham, Switzerland, 2017; pp. 123–149. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; LeCleir, G.R.; Bullerjahn, G.S.; McKay, R.M.; Saxton, M.A.; Twiss, M.R.; Bourbonniere, R.A. Seasonal changes in microbial community structure and activity imply winter production is linked to summer hypoxia in a large lake. FEMS Microbiol. Ecol. 2013, 87, 475–485. [Google Scholar] [CrossRef]
- Loreau, M.; Naeem, S.; Inchausti, P.; Bengtsson, J.; Grime, J.P.; Hector, A.; Hooper, D.U.; Huston, M.A.; Raffaelli, D.; Schmid, B.; et al. Biodiversity and Ecosystem Functioning: Current Knowledge and Future Challenges. Science 2001, 294, 804–808. [Google Scholar] [CrossRef] [Green Version]
- Price, J.; Ledford, S.H.; Ryan, M.O.; Toran, L.; Sales, C.M. Wastewater treatment plant effluent introduces recoverable shifts in microbial community composition in receiving streams. Sci. Total Environ. 2018, 613–614, 1104–1116. [Google Scholar] [CrossRef]
- Thomas, C.D.; Cameron, A.; Green, R.E.; Bakkenes, M.; Beaumont, L.J.; Collingham, Y.C.; Erasmus, B.F.N.; de Siqueira, M.F.; Grainger, A.; Hannah, L.; et al. Extinction risk from climate change. Nature 2004, 427, 145–148. [Google Scholar] [CrossRef]
- Trick, J.K.; Stuart, M.; Reeder, S. Contaminated groundwater sampling and quality control of water analyses. In Environmental Geochemistry: Site Characterization, Data Analysis and Case Histories. Candice Janco; Elsevier: Amsterdam, The Netherlands, 2018; pp. 25–45. [Google Scholar] [CrossRef] [Green Version]
- Logares, R.; Lindström, E.; Langenheder, S.; Logue, J.B.; Paterson, H.; Laybourn-Parry, J.; Rengefors, K.; Tranvik, L.J.; Bertilsson, S. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J. 2013, 7, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Malele, I.; Nyingilili, H.; Lyaruu, E.; Tauzin, M.; Ollivier, B.B.; Cayol, J.-L.; Fardeau, M.-L.; Geiger, A.; Malele, I.; Nyingilili, H.; et al. Bacterial diversity obtained by culturable approaches in the gut of Glossina pallidipes population from a non sleeping sickness focus in Tanzania: Preliminary results. BMC Microbiol. 2018, 18, 164. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xiang, P.; Duan, Z.; Fu, Z.; Zhang, L.; Zhang, Z. Electricity generation, salinity, COD removal and anodic biofilm microbial community vary with different anode CODs in a microbial desalination cell for high-salinity mustard tuber wastewater treatment. RSC Adv. 2019, 9, 25189–25198. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.H.; Guber, A.K.; Sadeghi, A.M.; Karns, J.S.; van Kessel, J.S.; Shelton, D.R.; Pachepsky, Y.A.; McCarty, G. Loss of bioactive phosphorus and enteric bacteria in runoff from dairy manure applied to sod. Soil Sci. 2008, 173, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Ren, M.; Xie, E.; Ding, A.; Liu, Y.; Deng, S.; Zhang, D. Roles of Phosphorus Sources in Microbial Community Assembly for the Removal of Organic Matters and Ammonia in Activated Sludge. Front. Microbiol. 2019, 10, 1023. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zheng, Y.; Teng, J.; Song, J.; Wang, X.; Zhao, Q. The seasonal variation of microbial communities in drinking water sources in Shanghai. J. Clean. Prod. 2020, 265, 121604. [Google Scholar] [CrossRef]
- Guan, Y.; Jia, J.; Wu, L.; Xue, X.; Zhang, G.; Wang, Z. Analysis of Bacterial Community Characteristics, Abundance of Antibiotics and Antibiotic Resistance Genes Along a Pollution Gradient of Ba River in Xi’an, China. Front. Microbiol. 2018, 9, 3191. [Google Scholar] [CrossRef] [Green Version]
- Visca, A.; Caracciolo, A.B.; Grenni, P.; Rolando, L.; Mariani, L.; Rauseo, J.; Spataro, F.; Monostory, K.; Sperlagh, B.; Patrolecco, L. Legacy and Emerging Pollutants in an Urban River Stretch and Effects on the Bacterioplankton Community. Water 2021, 13, 3402. [Google Scholar] [CrossRef]
- Perreten, V.; Vorlet-Fawer, L.; Slickers, P.; Ehricht, R.; Kuhnert, P.; Frey, J. Microarray-Based Detection of 90 Antibiotic Resistance Genes of Gram-Positive Bacteria. J. Clin. Microbiol. 2005, 43, 2291–2302. [Google Scholar] [CrossRef] [Green Version]
- Barati, A.; Ghaderpour, A.; Chew, L.L.; Bong, C.W.; Thong, K.L.; Chong, V.C.; Chai, L.C. Isolation and Characterization of Aquatic-Borne Klebsiella pneumoniae from Tropical Estuaries in Malaysia. Int. J. Environ. Res. Public Health 2016, 13, 426. [Google Scholar] [CrossRef] [Green Version]
- Ghaderpour, A.; Nasori, K.N.M.; Chew, L.L.; Chong, V.; Thong, K.L.; Chai, L.C. Detection of multiple potentially pathogenic bacteria in Matang mangrove estuaries, Malaysia. Mar. Pollut. Bull. 2014, 83, 324–330. [Google Scholar] [CrossRef]
- Siu, L.K.; Fung, C.-P.; Chang, F.-Y.; Lee, N.; Yeh, K.-M.; Koh, T.H.; Ip, M. Molecular Typing and Virulence Analysis of Serotype K1 Klebsiella pneumoniae Strains Isolated from Liver Abscess Patients and Stool Samples from Noninfectious Subjects in Hong Kong, Singapore, and Taiwan. J. Clin. Microbiol. 2011, 49, 3761–3765. [Google Scholar] [CrossRef] [Green Version]
- Wyres, K.; Holt, K. Klebsiella pneumoniae as a key trafficker of drug resistance genes from environmental to clinically important bacteria. Curr. Opin. Microbiol. 2018, 45, 131–139. [Google Scholar] [CrossRef]
- Flanagan, J.L.; Brodie, E.L.; Weng, L.; Lynch, S.V.; Garcia, O.; Brown, R.; Hugenholtz, P.; DeSantis, T.Z.; Andersen, G.L.; Wiener-Kronish, J.P.; et al. Loss of Bacterial Diversity during Antibiotic Treatment of Intubated Patients Colonized with Pseudomonas aeruginosa. J. Clin. Microbiol. 2007, 45, 1954–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Pang, W.; Dou, C.; Yin, D. Sulfamethoxazole and COD increase abundance of sulfonamide resistance genes and change bacterial community structures within sequencing batch reactors. Chemosphere 2017, 175, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ogo, M.; Koike, T.; Takada, H.; Newman, B. Sulfonamide and tetracycline resistance genes in total- and culturable-bacterial assemblages in South African aquatic environments. Front. Microbiol. 2015, 6, 796. [Google Scholar] [CrossRef] [PubMed]
- Björkman, J.; Andersson, D.I. The cost of antibiotic resistance from a bacterial perspective. Drug Resist. Updates 2000, 3, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Schulz zur Wiesch, P.; Engelstädter, J.; Bonhoeffer, S. Compensation of Fitness Costs and Reversibility of Antibiotic Resistance Mutations. Antimicrob. Agents Chemother. 2010, 54, 2085–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majcherczyk, P.A.; Barblan, J.-L.; Moreillon, P.; Entenza, J.M. Development of glycopeptide-intermediate resistance by Staphylococcus aureus leads to attenuated infectivity in a rat model of endocarditis. Microb. Pathog. 2008, 45, 408–414. [Google Scholar] [CrossRef]
- Andersson, D.I. The biological cost of mutational antibiotic resistance: Any practical conclusions? Curr. Opin. Microbiol. 2006, 9, 461–465. [Google Scholar] [CrossRef]
- Zhang, Q.; Sahin, O.; McDermott, P.F.; Payot, S. Fitness of antimicrobial-resistant Campylobacter and Salmonella. Microbes Infect. 2006, 8, 1972–1978. [Google Scholar] [CrossRef] [Green Version]
- Randall, L.P.; Bagnall, M.C.; Karatzas, K.A.; Coldham, N.C.; Piddock, L.J.V.; Woodward, M.J. Fitness and dissemination of disinfectant-selected multiple-antibiotic-resistant (MAR) strains of Salmonella enterica serovar Typhimurium in chickens. J. Antimicrob. Chemother. 2008, 61, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I. Persistence of antibiotic resistant bacteria. Curr. Opin. Microbiol. 2003, 6, 452–456. [Google Scholar] [CrossRef]
- Lipsitch, M. The rise and fall of antimicrobial resistance. Trends Microbiol. 2001, 9, 438–444. [Google Scholar] [CrossRef]
- Maisnier-Patin, S.; Andersson, D.I. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004, 155, 360–369. [Google Scholar] [CrossRef]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef]
- Fitzpatrick, D.; Walsh, F. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol. Ecol. 2016, 92, fiv168. [Google Scholar] [CrossRef]
- Bloomfield, S.F.; Stewart, G.S.A.B.; Dodd, C.E.R.; Booth, I.R.; Power, E.G.M. The viable but non-culturable phenomenon explained? Microbiology 1998, 144, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Takami, H.; Inoue, A.; Fuji, F.; Horikoshi, K. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol. Lett. 1997, 152, 279–285. [Google Scholar] [CrossRef]
- Azizian, M.; Pakzad, I.; Arabi, H.; Nasrollahi, A.; Hosainzadegan, H.; Azizi Jalilian, F.; Taherikalani, M.; Sadeghifard, N.; Samadi, N.; Nasser, A. Prevalence of dfr, int and sul Genes in Cotrimoxazole Resistance Klebsiella pneumoniae Isolated from Two Hospitals of Iran. J. Pure Appl. Microbiol. 2014, 8, 2655–2658. [Google Scholar]
- Hoa, P.T.P.; Managaki, S.; Nakada, N.; Takada, H.; Shimizu, A.; Anh, D.H.; Viet, P.H.; Suzuki, S. Antibiotic contamination and occurrence of antibiotic-resistant bacteria in aquatic environments of northern Vietnam. Sci. Total Environ. 2011, 409, 2894–2901. [Google Scholar] [CrossRef]
- Khamesipour, F.; Tajbakhsh, E. Analyzed the Genotypic and Phenotypic Antibiotic Resistance Patterns of Klebsiella pneumoniae Isolated from Clinical Samples in Iran. Biomed. Res. 2016, 27, 1017–1026. [Google Scholar]
- Liu, Y.; Cheng, D.; Xue, J.; Weaver, L.; Wakelin, S.A.; Feng, Y.; Li, Z. Changes in microbial community structure during pig manure composting and its relationship to the fate of antibiotics and antibiotic resistance genes. J. Hazard. Mater. 2020, 389, 122082. [Google Scholar] [CrossRef]
- Argudín, M.A.; Deplano, A.; Meghraoui, A.; Dodémont, M.; Heinrichs, A.; Denis, O.; Nonhoff, C.; Roisin, S. Bacteria from Animals as a Pool of Antimicrobial Resistance Genes. Antibiotics 2017, 6, 12. [Google Scholar] [CrossRef]
- Tsubakishita, S.; Kuwahara-Arai, K.; Baba, T.; Hiramatsu, K. Staphylococcal Cassette Chromosome mec -Like Element in Macrococcus caseolyticus. Antimicrob. Agents Chemother. 2010, 54, 1469–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, B.; Lemmer, H.; Horn, H.; Müller, E. Characterization of pure cultures isolated from sulfamethoxazole-acclimated activated sludge with respect to taxonomic identification and sulfamethoxazole biodegradation potential. BMC Microbiol. 2013, 13, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricken, B.; Kolvenbach, B.A.; Bergesch, C.; Benndorf, D.; Kroll, K.; Strnad, H.; Vlček, Č.; Adaixo, R.; Hammes, F.; Shahgaldian, P.; et al. FMNH2-dependent monooxygenases initiate catabolism of sulfonamides in Microbacterium sp. strain BR1 subsisting on sulfonamide antibiotics. Sci. Rep. 2017, 7, 15783. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Moore, I.F.; Koteva, K.; Bareich, D.C.; Hughes, D.W.; Wright, G. TetX Is a Flavin-dependent Monooxygenase Conferring Resistance to Tetracycline Antibiotics. J. Biol. Chem. 2004, 279, 52346–52352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-W.; Thawng, C.N.; Lee, K.; Wellington, E.M.; Cha, C.-J. A novel sulfonamide resistance mechanism by two-component flavin-dependent monooxygenase system in sulfonamide-degrading actinobacteria. Environ. Int. 2019, 127, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Masco, L.; Van Hoorde, K.; De Brandt, E.; Swings, J.; Huys, G. Antimicrobial susceptibility of Bifidobacterium strains from humans, animals and probiotic products. J. Antimicrob. Chemother. 2006, 58, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Beattie, T.K.; Knapp, C.W. Relationship between antibiotic- and disinfectant-resistance profiles in bacteria harvested from tap water. Chemosphere 2016, 152, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Vaz-Moreira, I.; Nunes, O.; Manaia, C.M. Diversity and Antibiotic Resistance Patterns of Sphingomonadaceae Isolates from Drinking Water. Appl. Environ. Microbiol. 2011, 77, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Ruan, X.; Xie, X.; Yan, Z. Antibiotic resistome profile based on metagenomics in raw surface drinking water source and the influence of environmental factor: A case study in Huaihe River Basin, China. Environ. Pollut. 2019, 248, 438–447. [Google Scholar] [CrossRef]
- Du, B.; Yang, Q.; Wang, R.; Wang, R.; Wang, Q.; Xin, Y. Evolution of Antibiotic Resistance and the Relationship between the Antibiotic Resistance Genes and Microbial Compositions under Long-Term Exposure to Tetracycline and Sulfamethoxazole. Int. J. Environ. Res. Public Health 2019, 16, 4681. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Sanchez, P.; Martinez, J.L. Environmental selection of antibiotic resistance genes. Minireview. Environ. Microbiol. 2001, 3, 1–9. [Google Scholar] [CrossRef]
- Van Hoek, A.H.; Veenman, C.; van Overbeek, W.M.; Lynch, G.; Husman, A.M.D.R.; Blaak, H. Prevalence and characterization of ESBL- and AmpC-producing Enterobacteriaceae on retail vegetables. Int. J. Food Microbiol. 2015, 204, 1–8. [Google Scholar] [CrossRef]
- Narciso-Da-Rocha, C.; Manaia, C.M. Multidrug resistance phenotypes are widespread over different bacterial taxonomic groups thriving in surface water. Sci. Total Environ. 2016, 563–564, 1–9. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W.; Lin, H.; Wang, W.; Du, L.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Primer | Sequence | Size (bp) | Reference |
|---|---|---|---|---|
| 16S rDNA | 27F | 5′-AGAGTTTGATCMTGGCTCAG-3′ | 1500 | [4] |
| 1492R | 5′-GGTTACCTTGTTACGACTT-3′ | |||
| sul1 | Sul1F | 5′-CGGCGTGGGCTACCTGAACG-3′ | 433 | [33] |
| Sul1R | 5′-GCCGATCGCGTGAAGTTCCG-3′ | |||
| sul2 | Sul2F | 5′-GCGCTCAAGGCAGATGGCATT-3′ | 293 | [33] |
| Sul2R | 5′-GCGTTTGATACCGGCACCCGT-3′ | |||
| sul3 | Sul3F | 5′-CCCATACCCGGATCAAGAATAA-3′ | 143 | [34] |
| Sul3R | 5′-CAGCGAATTGGTGCAGCTACTA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lye, Y.-L.; Chai, L.-C.; Lee, C.-W.; Suzuki, S.; Bong, C.-W. Microbial Community Structure and Bacterial Lineages Associated with Sulfonamides Resistance in Anthropogenic Impacted Larut River. Water 2022, 14, 1018. https://doi.org/10.3390/w14071018
Lye Y-L, Chai L-C, Lee C-W, Suzuki S, Bong C-W. Microbial Community Structure and Bacterial Lineages Associated with Sulfonamides Resistance in Anthropogenic Impacted Larut River. Water. 2022; 14(7):1018. https://doi.org/10.3390/w14071018
Chicago/Turabian StyleLye, Ying-Ling, Lay-Ching Chai, Choon-Weng Lee, Satoru Suzuki, and Chui-Wei Bong. 2022. "Microbial Community Structure and Bacterial Lineages Associated with Sulfonamides Resistance in Anthropogenic Impacted Larut River" Water 14, no. 7: 1018. https://doi.org/10.3390/w14071018