
Metatranscriptome Analysis of Sympatric Bee Species Identifies Bee Virus Variants and a New Virus, Andrena-Associated Bee Virus-1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
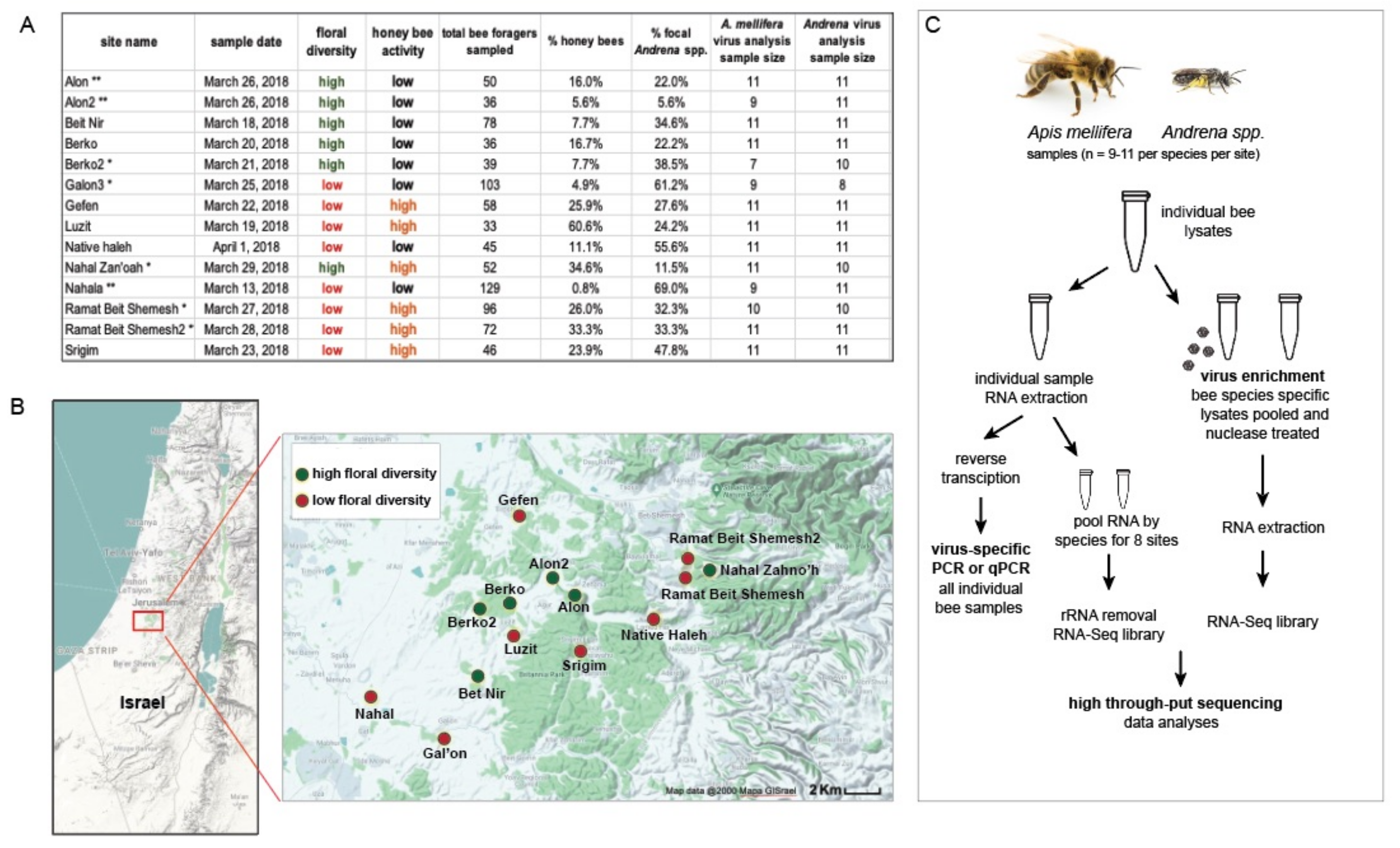
2.1. Study Region and Survey Design
Virus Analysis
2.2. RNA Extraction
2.3. RNAseq Library Preparation and Sequencing
2.4. Apis mellifera (Honey Bee) Genome Alignment
2.5. HoloBee Database Expansion (March 2019)
2.6. RNA Sequencing and Analyses
2.7. Reverse Transcription/cDNA Synthesis
2.8. Polymerase Chain Reaction (PCR)
2.9. Quantitative Polymerase Chain Reaction (qPCR)
2.10. Statistical Analysis of qPCR
2.11. Negative Strand-Specific PCR
2.12. Rapid Amplification of cDNA Ends
2.13. Honey Bee Pupal Cell Cultures and AnBV-1 Infection
2.14. Andrena-associated bee virus-1 (AnBV-1) Genome and Phylogenetic Analyses
2.15. Statistical Modeling of AnBV-1 Prevalence in the Field
3. Results and Discussion
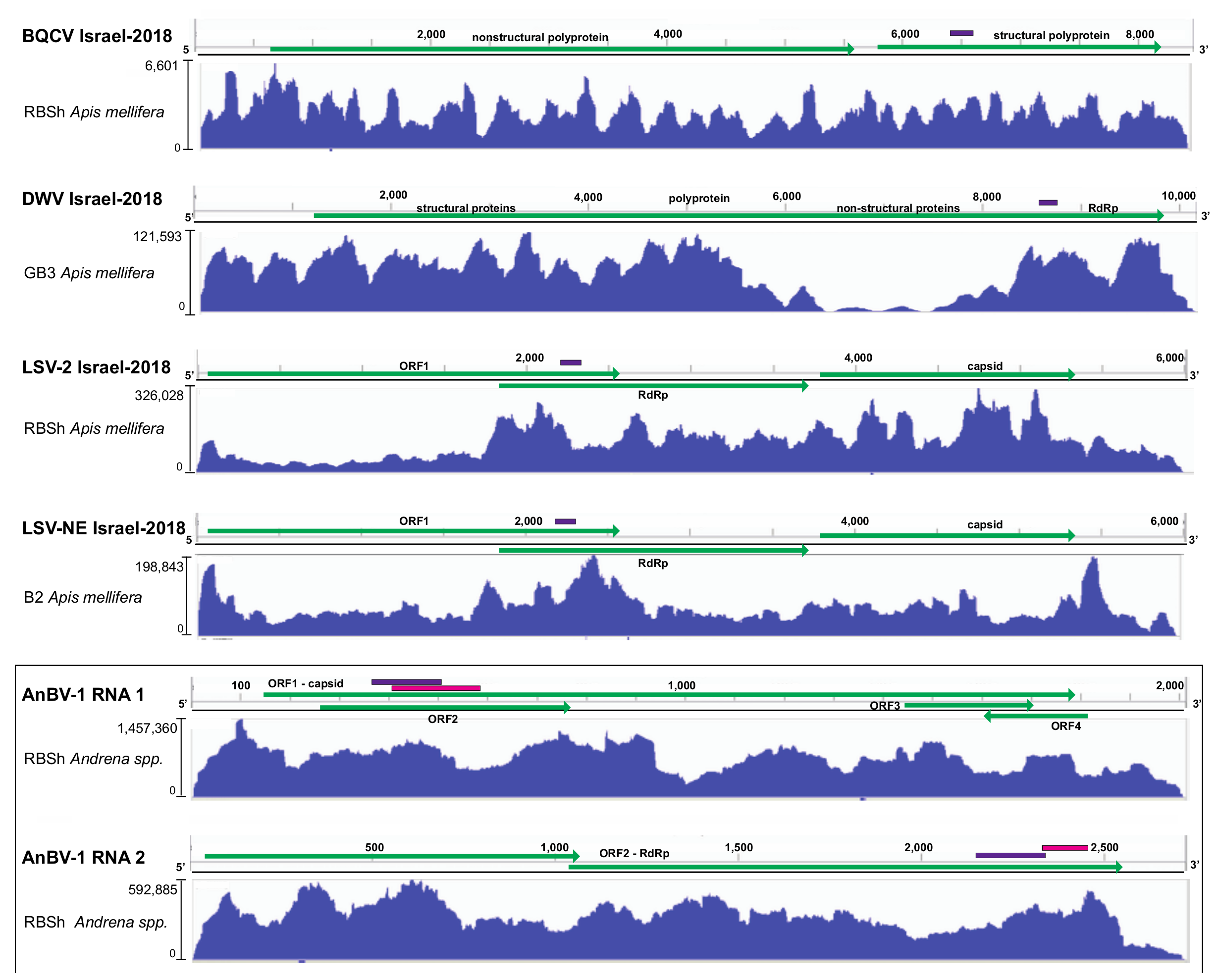
3.1. Metatranscriptome Assessment of Viruses in Sympatric Bee Species
3.2. Sequence Variants of Bee-Infecting Viruses: Black Queen Cell Virus, Deformed Wing Virus, and Members of the Lake Sinai Virus Group
3.2.1. Black queen cell virus
3.2.2. Lake Sinai virus
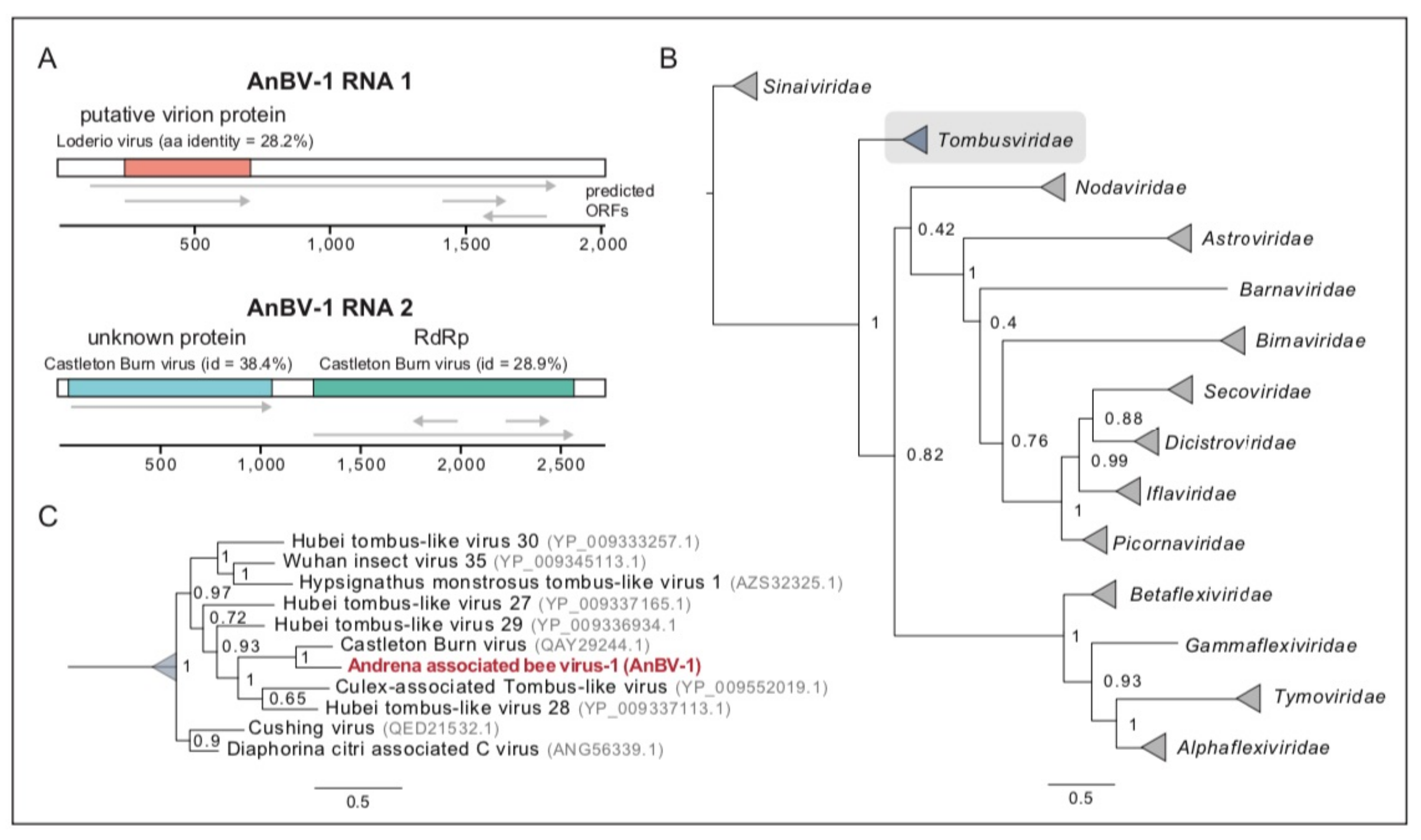
3.3. Novel Bipartite Positive-Sense RNA Virus Identified in Andrena Mining Bees
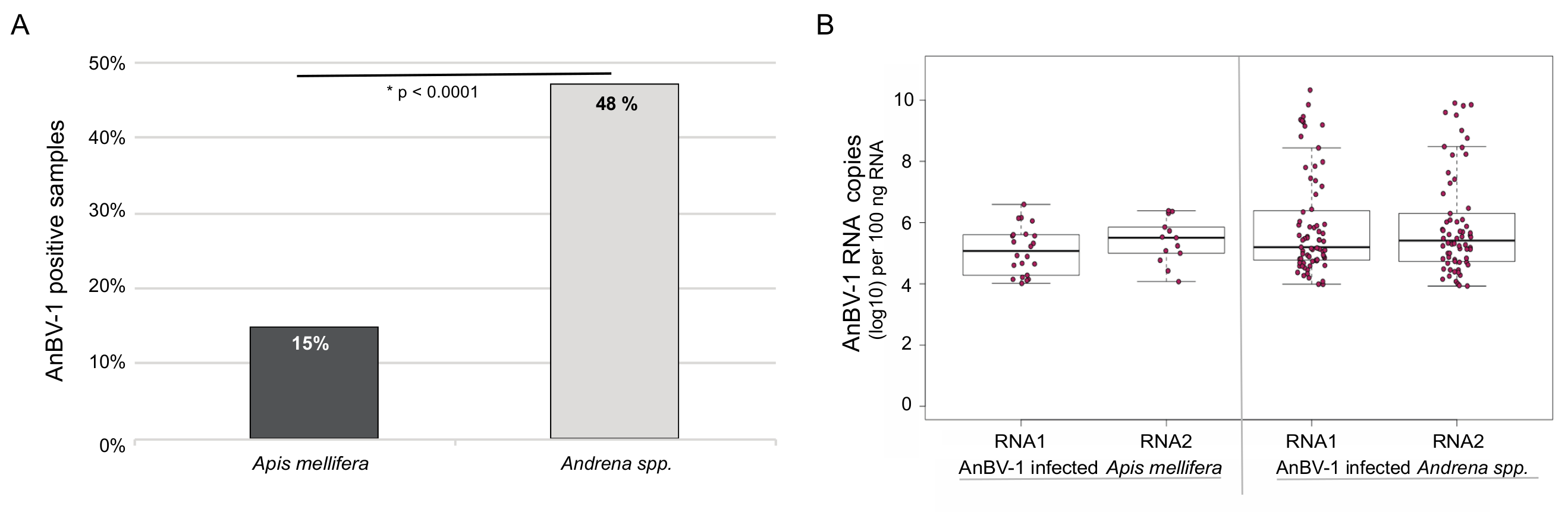
3.4. Andrena-associated bee virus-1 (AnBV-1) is More Prevalent in Mining Bees
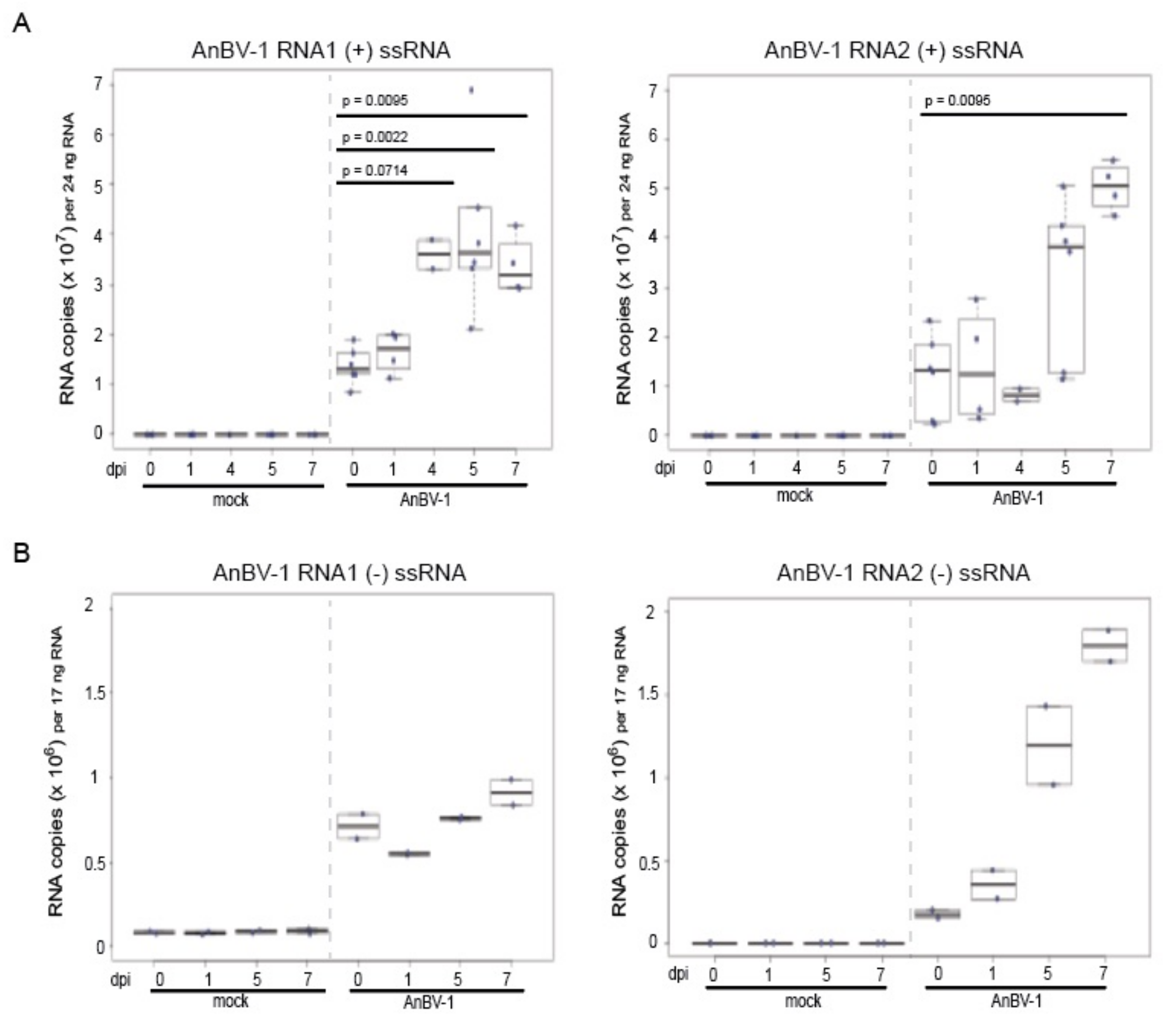
3.5. AnBV-1 Replicates in Primary Honey Bee Pupal Cell Cultures
3.6. Sequence Analyses Indicate Virus Transmission between Sympatric Bee Species
3.7. The Probability of AnBV-1 Infection in Honey Bees Is Modulated by the Floral Community
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ollerton, J.; Winfree, R.; Tarrant, S. How many flowering plants are pollinated by animals? Oikos 2011, 120, 321–326. [Google Scholar] [CrossRef]
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Michener, C.D. The Bees of the World, 2nd ed.; John Hopkins University Press: Baltimore, MD, USA; London, UK, 2007. [Google Scholar]
- Cariveau, D.P.; Nayak, G.K.; Bartomeus, I.; Zientek, J.; Ascher, J.S.; Gibbs, J.; Winfree, R. The Allometry of Bee Proboscis Length and Its Uses in Ecology. PLoS ONE 2016, 11, e0151482. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, C.; Debano, S.J.; Thorp, R.W.; Rao, S.; Stephen, W.P. Investigating temporal patterns of a native bee community in a remnant North American bunchgrass prairie using blue vane traps. J. Insect Sci. 2012, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Gathmann, A.; Tscharntke, T. Foraging ranges of solitary bees. J. Anim. Ecol. 2002, 71, 757–764. [Google Scholar] [CrossRef]
- Wagner, D.L. Insect Declines in the Anthropocene. Annu. Rev. Entomol. 2020, 65, 457–480. [Google Scholar] [CrossRef] [Green Version]
- Van Klink, R.; Bowler, D.E.; Gongalsky, K.B.; Swengel, A.B.; Gentile, A.; Chase, J.M. Meta-analysis reveals declines in terrestrial but increases in freshwater insect abundances. Science 2020, 368, 417. [Google Scholar] [CrossRef]
- Duchenne, F.; Thebault, E.; Michez, D.; Gerard, M.; Devaux, C.; Rasmont, P.; Vereecken, N.J.; Fontaine, C. Long-term effects of global change on occupancy and flight period of wild bees in Belgium. Glob. Chang. Biol. 2020, 26, 6753–6766. [Google Scholar] [CrossRef]
- Goulson, D.; Nicholls, E.; Botias, C.; Rotheray, E.L. Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 2015, 347, 1255957. [Google Scholar] [CrossRef]
- Mathiasson, M.; Rehan, S. Status changes in the wild bees of north-eastern North America over 125 years revealed through museum specimens. Insect Conserv. Divers. 2019, 12, 278–288. [Google Scholar] [CrossRef]
- Goulson, D.; Lye, G.C.; Darvill, B. Decline and Conservation of Bumble Bees. Annu. Rev. Entomol. 2008, 53, 191–208. [Google Scholar] [CrossRef]
- Cameron, S.A.; Lozier, J.D.; Strange, J.P.; Koch, J.B.; Cordes, N.; Solter, L.F.; Griswold, T.L. Patterns of widespread decline in North American bumble bees. Proc. Natl. Acad. Sci. USA 2011, 108, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Arbetman, M.P.; Gleiser, G.; Morales, C.L.; Williams, P.; Aizen, M.A. Global decline of bumblebees is phylogenetically structured and inversely related to species range size and pathogen incidence. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170204. [Google Scholar] [CrossRef] [Green Version]
- Aizen, M.A.; Harder, L.D. The Global Stock of Domesticated Honey Bees Is Growing Slower Than Agricultural Demand for Pollination. Curr. Biol. 2009, 19, 915–918. [Google Scholar] [CrossRef] [Green Version]
- van Engelsdorp, D.; Meixner, M.D. A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 2010, 103, S80–S95. [Google Scholar] [CrossRef] [PubMed]
- Russo, L. Positive and Negative Impacts of Non-Native Bee Species around the World. Insects 2016, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Stork, N.E. How Many Species of Insects and Other Terrestrial Arthropods Are There on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Langstroth, L. Langstroth on the Hive and the Honey Bee; Xist Publishing: Rosenberg, TX, USA, 2014. [Google Scholar]
- Beekman, M.; Ratnieks, F.L.W. Long-Range Foraging by the Honey-Bee, Apis mellifera L. Funct. Ecol. 2000, 14, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.K.; Seeley, T.D. Foraging Strategy of Honeybee Colonies in a Temperate Deciduous Forest. Ecology 1982, 63, 1790–1801. [Google Scholar] [CrossRef]
- Couvillon, M.J.; Pearce, F.C.R.; Accleton, C.; Fensome, K.A.; Quah, S.K.L.; Taylor, E.L.; Ratnieks, F.L.W. Honey bee foraging distance depends on month and forage type. Apidologie 2015, 46, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Soroker, V.; Hetzroni, A.; Yakobson, B.; David, D.; David, A.; Voet, H.; Slabezki, Y.; Efrat, H.; Levski, S.; Kamer, Y.; et al. Evaluation of colony losses in Israel in relation to the incidence of pathogens and pests. Apidologie 2011, 42. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.V.; Steinhauer, N.; Rennich, K.; Wilson, M.E.; Tarpy, D.R.; Caron, D.M.; Rose, R.; Delaplane, K.S.; Baylis, K.; Lengerich, E.J.; et al. A national survey of managed honey bee 2013–2014 annual colony losses in the USA. Apidologie 2015, 46, 292–305. [Google Scholar] [CrossRef] [Green Version]
- Spleen, A.M.; Lengerich, E.J.; Rennich, K.; Caron, D.; Rose, R.; Pettis, J.S.; Henson, M.; Wilkes, J.T.; Wilson, M.; Stitzinger, J.; et al. A national survey of managed honey bee 2011–2012 winter colony losses in the United States: Results from the Bee Informed Partnership. J. Apic. Res. 2015, 52, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Steinhauer, N.; van Engelsdorp, D. Using Epidemiological Methods to Improve Honey Bee Colony Health. In Beekeeping—From Science to Practice; Springer International Publishing: Cham, Switzerland, 2017; Volume 19, pp. 125–142. [Google Scholar]
- Steinhauer, N.A.; Rennich, K.; Wilson, M.E.; Caron, D.M.; Lengerich, E.J.; Pettis, J.S.; Rose, R.; Skinner, J.A.; Tarpy, D.R.; Wilkes, J.T.; et al. A national survey of managed honey bee 2012–2013 annual colony losses in the USA: Results from the Bee Informed Partnership. J. Apic. Res. 2015, 53, 1–18. [Google Scholar] [CrossRef]
- Traynor, K.S.; Rennich, K.; Forsgren, E.; Rose, R.; Pettis, J.; Kunkel, G.; Madella, S.; Evans, J.; Lopez, D.; vanEngelsdorp, D. Multiyear survey targeting disease incidence in US honey bees. Apidologie 2016, 47, 325–347. [Google Scholar] [CrossRef] [Green Version]
- van Engelsdorp, D.; Caron, D.; Hayes, J.; Underwood, R.; Henson, M.; Rennich, K.; Spleen, A.; Andree, M.; Snyder, R.; Lee, K.; et al. A national survey of managed honey bee 2010–11 winter colony losses in the USA: Results from the Bee Informed Partnership. J. Apic. Res. 2015, 51, 115–124. [Google Scholar] [CrossRef] [Green Version]
- van Engelsdorp, D.; Hayes, J.; Underwood, R.M.; Pettis, J. A Survey of Honey Bee Colony Losses in the U.S. Fall 2007 to Spring 2008. PLoS ONE 2008, 3, e4071. [Google Scholar]
- Bruckner, S.; Steinhauer, N.; Aurell, S.D.; Caron, D.M.; Ellis, J.D.; Fauvel, A.M.; Kulhanek, K.; McArt, S.; Mullen, E.; Rangel, J.; et al. 2018–2019 Honey Bee Colony Losses in the United States: Preliminary Results; The Bee Informed Partnership: Bethesda, MD, USA, 2020. [Google Scholar]
- Seitz, N.; Traynor, K.S.; Steinhauer, N.; Rennich, K.; Wilson, M.E.; Ellis, J.D.; Rose, R.; Tarpy, D.R.; Sagili, R.R.; Caron, D.M.; et al. A national survey of managed honey bee 2014–2015 annual colony losses in the USA. J. Apic. Res. 2015, 54, 292–304. [Google Scholar] [CrossRef]
- Kulhanek, K.; Steinhauer, N.; Rennich, K.; Caron, D.M.; Sagili, R.R.; Pettis, J.S.; Ellis, J.D.; Wilson, M.E.; Wilkes, J.T.; Tarpy, D.R.; et al. A national survey of managed honey bee 2015–2016 annual colony losses in the USA. J. Apic. Res. 2017, 56, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Ascher, J.S. Pickering Discover Life Bee Species Guide and World Checklist (Hymenoptera: Apoidea: Anthophila). Available online: http://www.discoverlife.org/mp/20q?guide=Apoidea_species (accessed on 12 January 2020).
- Pisanty, G.; Richter, R.; Martin, T.; Dettman, J.; Cardinal, S. Molecular phylogeny and historical biogeography of andrenine bees (Hymenoptera: Andrenidae). bioRxiv 2020. [Google Scholar] [CrossRef]
- Paxton, R.J.; Thoren, P.A.; Tengo, J.; Estoup, A.; Pamilo, P. Mating structure and nestmate relatedness in a communal bee, Andrena jacobi (Hymenoptera, Andrenidae), using microsatellites. Mol. Ecol. 1996, 5, 511–519. [Google Scholar] [CrossRef]
- Michener, C.D. The Social Behavior of the Bees: A Comparative Study; Belknap Pr.: Cambridge, MA, USA, 1974. [Google Scholar]
- Wood, T.J.; Roberts, S.P.M. An assessment of historical and contemporary diet breadth in polylectic Andrena bee species. Biol. Conserv. 2017, 215, 72–80. [Google Scholar] [CrossRef]
- Larkin, L.L.; Neff, J.L.; Simpson, B.B. The evolution of a pollen diet: Host choice and diet breadth of Andrena bees (Hymenoptera: Andrenidae). Apidologie 2008, 39, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Cornman, R.S.; Tarpy, D.R.; Chen, Y.; Jeffreys, L.; Lopez, D.; Pettis, J.S.; vanEngelsdorp, D.; Evans, J.D. Pathogen Webs in Collapsing Honey Bee Colonies. PLoS ONE 2012, 7, e43562. [Google Scholar] [CrossRef] [Green Version]
- Brutscher, L.M.; McMenamin, A.J.; Flenniken, M.L. The Buzz about Honey Bee Viruses. PLoS Pathog. 2016, 12, e1005757. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Flenniken, M.L. Bee Viruses: Ecology, Pathogenicity, and Impacts. Annu. Rev. Entomol. 2019, 64, 205–226. [Google Scholar] [CrossRef]
- McMenamin, A.J.; Flenniken, M.L. Recently identified bee viruses and their impact on bee pollinators. Curr. Opin. Insect Sci. 2018, 26, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Ryabov, E.V. Invertebrate RNA virus diversity from a taxonomic point of view. J. Invertebr. Pathol. 2017, 147, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; et al. Diversity and Global Distribution of Viruses of the Western Honey Bee, Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef]
- Gauthier, L.; Cornman, S.; Hartmann, U.; Cousserans, F.; Evans, J.; de Miranda, J.; Neumann, P. The Apis mellifera Filamentous Virus Genome. Viruses 2015, 7, 3798–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, C.; Li, B.; Deng, S.; Chu, Y.; Diao, Q. Diagnosis and distribution of the Apis mellifera filamentous virus (AmFV) in honey bees (Apis mellifera) in China. Insectes Soc. 2017, 64, 597–603. [Google Scholar] [CrossRef]
- Galbraith, D.A.; Fuller, Z.; Brockmann, A.; Frazier, M.; Gikungu, M.W.; Kapheim, K.M.; Kerby, J.T.; Kocher, S.D.; Losyev, O.; Muli, E.; et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoonvaere, K.; Smagghe, G.; Francis, F.; de Graaf, D.C. Study of the Metatranscriptome of Eight Social and Solitary Wild Bee Species Reveals Novel Viruses and Bee Parasites. Front. Microbiol. 2018, 9, 177. [Google Scholar] [CrossRef]
- Ravoet, J.; De Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; de Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, A.G.; Hendrix, S.D.; Scavo, N.A.; Carrillo-Tripp, J.; Harris, M.A.; Wheelock, M.J.; O’Neal, M.E.; Toth, A.L. Honey Bee Viruses in Wild Bees: Viral Prevalence, Loads, and Experimental Inoculation. PLoS ONE 2016, 11, e0166190. [Google Scholar] [CrossRef] [PubMed]
- Alger, S.A.; Burnham, P.A.; Boncristiani, H.F.; Brody, A.K. RNA virus spillover from managed honeybees (Apis mellifera) to wild bumblebees (Bombus spp.). PLoS ONE 2019, 14, e0217822. [Google Scholar] [CrossRef] [Green Version]
- Alger, S.A.; Burnham, P.A.; Brody, A.K. Flowers as viral hot spots: Honey bees (Apis mellifera) unevenly deposit viruses across plant species. PLoS ONE 2019, 14, e0221800. [Google Scholar] [CrossRef]
- Tehel, A.; Brown, M.J.; Paxton, R.J. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; He, S.Y.; Evans, J.D.; Pettis, J.S.; Yin, G.F.; Chen, Y.P. New evidence that deformed wing virus and black queen cell virus are multi-host pathogens. J. Invertebr. Pathol. 2012, 109, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; van Engelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA Viruses in Hymenopteran Pollinators: Evidence of Inter-Taxa Virus Transmission via Pollen and Potential Impact on Non-Apis Hymenopteran Species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef]
- Radzevičiūtė, R.; Theodorou, P.; Husemann, M.; Japoshvili, G.; Kirkitadze, G.; Zhusupbaeva, A.; Paxton, R.J. Replication of honey bee-associated RNA viruses across multiple bee species in apple orchards of Georgia, Germany and Kyrgyzstan. J. Invertebr. Pathol. 2017, 146, 14–23. [Google Scholar] [CrossRef] [Green Version]
- de Miranda, J.; Cornman, R.; Evans, J.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome Characterization, Prevalence and Distribution of a Macula-Like Virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 3586–3602. [Google Scholar] [CrossRef] [Green Version]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blacquiere, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A Diverse Range of Novel RNA Viruses in Geographically Distinct Honey Bee Populations. J. Virol. 2017, 91, e00158-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, S.; Galbraith, D.; Sela, N.; Erez, T.; Grozinger, C.M.; Chejanovsky, N. Presence of Apis Rhabdovirus-1 in Populations of Pollinators and Their Parasites from Two Continents. Front. Microbiol. 2017, 8, 2482. [Google Scholar] [CrossRef] [Green Version]
- Runckel, C.; Flenniken, M.L.; Engel, J.C.; Ruby, J.G.; Ganem, D.; Andino, R.; DeRisi, J.L. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 2011, 6, e20656. [Google Scholar] [CrossRef] [Green Version]
- Levin, S.; Sela, N.; Chejanovsky, N. Two novel viruses associated with the Apis mellifera pathogenic mite Varroa destructor. Sci. Rep. 2016, 6, 37710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, S.; Sela, N.; Erez, T.; Nestel, D.; Pettis, J.; Neumann, P.; Chejanovsky, N. New Viruses from the Ectoparasite Mite Varroa destructor Infesting Apis mellifera and Apis cerana. Viruses 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.N.; Shepherd, T.F.; Rangel, J. The detection of honey bee (Apis mellifera)-associated viruses in ants. Sci. Rep. 2020, 10, 2923. [Google Scholar] [CrossRef] [Green Version]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 2016, 6, 1304. [Google Scholar] [CrossRef]
- Chen, Y.P.; Siede, R. Honey Bee Viruses. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2007; Volume 70, pp. 33–80. [Google Scholar]
- Bowen-Walker, P.L.; Martin, S.J.; Gunn, A. The transmission of deformed wing virus between honeybees (Apis mellifera L.) by the ectoparasitic mite Varroa jacobsoni Oud. J. Invertebr. Pathol. 1999, 73, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Nazzi, F.; Brown, S.P.; Annoscia, D.; Del Piccolo, F.; Di Prisco, G.; Varricchio, P.; Della Vedova, G.; Cattonaro, F.; Caprio, E.; Pennacchio, F. Synergistic Parasite-Pathogen Interactions Mediated by Host Immunity Can Drive the Collapse of Honeybee Colonies. PLoS Pathog. 2012, 8, e1002735. [Google Scholar] [CrossRef] [Green Version]
- Ongus, J.R. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef]
- Wilfert, L.; Long, G.; Leggett, H.C.; Schmid-Hempel, P.; Butlin, R.; Martin, S.J.M.; Boots, M. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 2016, 351, 594–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.P.; Pettis, J.S.; Corona, M.; Chen, W.P.; Li, C.J.; Spivak, M.; Visscher, P.K.; De Grandi-Hoffman, G.; Boncristiani, H.; Zhao, Y.; et al. Israeli Acute Paralysis Virus: Epidemiology, Pathogenesis and Implications for Honey Bee Health. PLoS Pathog. 2014, 10, e1004261. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pettis, J.S.; Evans, J.D.; Kramer, M.; Fedlaufer, M. Transmission of Kashmir bee virus by the ectoparasitic mite Varroa destructor. Apidologie 2004, 35, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Celle, O.; Blanchard, P.; Olivier, V.; Schurr, F.; Cougoule, N.; Faucon, J.-P.; Ribière, M. Detection of Chronic bee paralysis virus (CBPV) genome and its replicative RNA form in various hosts and possible ways of spread. Virus Res. 2008, 133, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyer, M.; Chen, Y.P.; Schäfer, M.O.; Pettis, J.; Neumann, P. Small hive beetle, Aethina tumida, as a potential biological vector of honeybee viruses. Apidologie 2009, 40, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Kwadha, C.A.; Ong’amo, G.O.; Ndegwa, P.N.; Raina, S.K.; Fombong, A.T. The Biology and Control of the Greater Wax Moth, Galleria mellonella. Insects 2017, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Traiyasut, P.; Mookhploy, W.; Kimura, K.; Yoshiyama, M.; Khongphinitbunjong, K.; Chantawannakul, P. First Detection of Honey Bee Viruses in Wax Moth. Chiang Mai J. Sci. 2016, 43, 695–698. [Google Scholar]
- Di Prisco, G.; Annoscia, D.; Margiotta, M.; Ferrara, R.; Varricchio, P.; Zanni, V.; Caprio, E.; Nazzi, F.; Pennacchio, F. A mutualistic symbiosis between a parasitic mite and a pathogenic virus undermines honey bee immunity and health. Proc. Natl. Acad. Sci. USA 2016, 113, 3203–3208. [Google Scholar] [CrossRef] [Green Version]
- Gisder, S.; Aumeier, P.; Genersch, E. Deformed wing virus: Replication and viral load in mites (Varroa destructor). J. Gen. Virol. 2009, 90, 463–467. [Google Scholar] [CrossRef]
- Martin, S.J.; Brettell, L.E. Deformed Wing Virus in Honeybees and Other Insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef]
- Yanez, O.; Piot, N.; Dalmon, A.; de Miranda, J.R.; Chantawannakul, P.; Panziera, D.; Amiri, E.; Smagghe, G.; Schroeder, D.; Chejanovsky, N. Bee Viruses: Routes of Infection in Hymenoptera. Front. Microbiol. 2020, 11, 943. [Google Scholar] [CrossRef]
- McArt, S.H.; Koch, H.; Irwin, R.E.; Adler, L.S. Arranging the bouquet of disease: Floral traits and the transmission of plant and animal pathogens. Ecol. Lett. 2014, 17, 624–636. [Google Scholar] [CrossRef]
- Koch, H.; Brown, M.J.F.; Stevenson, P.C. The role of disease in bee foraging ecology. Curr. Opin. Insect Sci. 2017, 21, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzei, M.; Carrozza, M.L.; Luisi, E.; Forzan, M.; Giusti, M.; Sagona, S.; Tolari, F.; Felicioli, A. Infectivity of DWV Associated to Flower Pollen: Experimental Evidence of a Horizontal Transmission Route. PLoS ONE 2014, 9, e113448. [Google Scholar]
- Gisder, S.; Genersch, E. Viruses of commercialized insect pollinators. J. Invertebr. Pathol. 2017, 147, 51–59. [Google Scholar] [CrossRef]
- Tapia-González, J.M.; Morfin, N.; Macías-Macías, J.O.; De la Mora, A.; Tapia-Rivera, J.C.; Ayala, R.; Contreras-Escareño, F.; Gashout, H.A.; Guzman-Novoa, E. Evidence of presence and replication of honey bee viruses among wild bee pollinators in subtropical environments. J. Invertebr. Pathol. 2019, 168, 107256. [Google Scholar] [CrossRef]
- Fürst, M.A.; McMahon, D.P.; Osborne, J.L.; Paxton, R.J.; Brown, M.J.F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 2014, 506, 364–366. [Google Scholar] [CrossRef]
- Gusachenko, O.N.; Woodford, L.; Balbirnie-Cumming, K.; Ryabov, E.V.; Evans, D.J. Evidence for and against deformed wing virus spillover from honey bees to bumble bees: A reverse genetic analysis. Sci. Rep. 2020, 10, 16847. [Google Scholar] [CrossRef] [PubMed]
- Glenny, W.; Cavigli, I.; Daughenbaugh, K.F.; Radford, R.; Kegley, S.E.; Flenniken, M.L. Honey bee (Apis mellifera) colony health and pathogen composition in migratory beekeeping operations involved in California almond pollination. PLoS ONE 2017, 12, e0182814. [Google Scholar] [CrossRef]
- Cavigli, I.; Daughenbaugh, K.F.; Martin, M.; Lerch, M.; Banner, K.; Garcia, E.; Brutscher, L.M.; Flenniken, M.L. Pathogen prevalence and abundance in honey bee colonies involved in almond pollination. Apidologie 2016, 47, 251–266. [Google Scholar] [CrossRef] [Green Version]
- Faurot-Daniels, C.; Glenny, W.; Daughenbaugh, K.F.; McMenamin, A.J.; Burkle, L.A.-O.; Flenniken, M.A.-O. Longitudinal monitoring of honey bee colonies reveals dynamic nature of virus abundance and indicates a negative impact of Lake Sinai virus 2 on colony health. PLoS ONE 2020, 15, e0237544. [Google Scholar] [CrossRef] [PubMed]
- Alger, S.A.; Burnham, P.A.; Lamas, Z.S.; Brody, A.K.; Richardson, L.L. Home sick: Impacts of migratory beekeeping on honey bee (Apis mellifera) pests, pathogens, and colony size. PeerJ 2018, 6, e5812. [Google Scholar] [CrossRef] [Green Version]
- Ricigliano, V.A.; Mott, B.M.; Floyd, A.S.; Copeland, D.C.; Carroll, M.J.; Anderson, K.E. Honey bees overwintering in a southern climate: Longitudinal effects of nutrition and queen age on colony-level molecular physiology and performance. Sci. Rep. 2018, 8, 10475. [Google Scholar] [CrossRef] [PubMed]
- van Engelsdorp, D.; Tarpy, D.R.; Lengerich, E.J.; Pettis, J.S. Idiopathic brood disease syndrome and queen events as precursors of colony mortality in migratory beekeeping operations in the eastern United States. Prev. Vet. Med. 2013, 108, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genersch, E.; von der Ohe, W.; Kaatz, H.; Schroeder, A.; Otten, C.; Büchler, R.; Berg, S.; Ritter, W.; Mühlen, W.; Gisder, S.; et al. The German bee monitoring project: A long term study to understand periodically high winter losses of honey bee colonies. Apidologie 2010, 41, 332–352. [Google Scholar] [CrossRef] [Green Version]
- Nazzi, F.; Pennacchio, F. Disentangling multiple interactions in the hive ecosystem. Trends Parasitol. 2014, 30, 556–561. [Google Scholar] [CrossRef]
- Deboutte, W.; Beller, L.; Yinda, C.K.; Shi, C.; Smets, L.; Vanmechelen, B.; Conceição-Neto, N.; Dallmeier, K.; Maes, P.; de Graaf, D.C.; et al. Hymenoptera associated eukaryotic virome lacks host specificity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pisanty, G.; Mandelik, Y. Profiling crop pollinators: Life history traits predict habitat use and crop visitation by Mediterranean wild bees. Ecol. Appl. 2015, 25, 742–752. [Google Scholar] [CrossRef]
- Pisanty, G.; Afik, O.; Wajnberg, E.; Mandelik, Y. Watermelon pollinators exhibit complementarity in both visitation rate and single-visit pollination efficiency. J. Appl. Ecol. 2016, 53, 360–370. [Google Scholar] [CrossRef]
- Evans, J.D.; Schwarz, R.; Childers, A. HoloBee Database v2016.1. Ag Data Commons. 2016. Available online: https://doi.org/10.15482/USDA.ADC/1255217 (accessed on 1 February 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Fau-Wysoker, A.; Wysoker, A.; Fau-Fennell, T.; Fennell, T.; Fau-Ruan, J.; Ruan, J.; Fau-Homer, N.; Homer, N.; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runckel, C.; DeRisi, J.; Flenniken, M.L. A draft genome of the honey bee trypanosomatid parasite Crithidia mellificae. PLoS ONE 2014, 9, e95057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.A.-O.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- de Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.E.; Chejanovsky, N.; Chen, Y.-P.; Gauthier, L.; Genersch, E.; de Graaf, D.C.; et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2015, 52, 1–56. [Google Scholar] [CrossRef] [Green Version]
- Ginzinger, D.G. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 2002, 30, 503–512. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: http://www.R-project.org/ (accessed on 9 January 2020).
- Daughenbaugh, K.F.; Martin, M.; Brutscher, L.M.; Cavigli, I.; Garcia, E.; Lavin, M.; Flenniken, M.L. Honey Bee Infecting Lake Sinai Viruses. Viruses 2015, 7, 3285–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, W.B. Medium for development of bee cell cultures (Apis mellifera: Hymenoptera: Apidae). In Vitro Cell. Dev. Biol. Anim. 2010, 46, 83–86. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Makhsous, N.; Shean, R.; Jerome, K.; Crawford, R. Genome Sequences of Viruses from Spiders around Seattle. 2020. Available online: https://www.genome.jp/virushostdb/1911102 (accessed on 10 January 2020).
- Katoh, K.; Kuma, K.; Fau-Toh, H.; Toh, H.; Fau-Miyata, T.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Gl Fau-Doallo, R.T.; Doallo, R.; Fau-Posada, D.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.D.A. FigTree Version 1.4.0. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 9 January 2020).
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Package ‘lme4’. Available online: http://dk.archive.ubuntu.com/pub/pub/cran/web/packages/lme4/lme4.pdf (accessed on 9 January 2020).
- Ben Bolker, R.D.C.T. bbmle: Tools for General Maximum Likelihood Estimation Methods and Functions for Fitting Maximum Likelihood Models in R. Available online: https://CRAN.R-project.org/package=bbmle (accessed on 9 January 2020).
- Hartig, F. DHARMa: Residual Diagnostics for Hierarchical (Multi-Level/Mixed) Regression Models. Available online: https://cran.r-project.org/web/packages/DHARMa/vignettes/DHARMa.html (accessed on 9 January 2020).
- Fortin, M.-J.; Dale, M.R.T. Spatial Analysis: A Guide for Ecologists; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Consortium, H.G.S. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; Jironkin, A.; Chandler, D.; Burroughs, N.; Evans, D.J.; Ryabov, E.V. Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J. Gen. Virol. 2010, 92, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Pettis, J.S.; Zhao, Y.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Li, R.; Zheng, H.; Huang, S.; Zhang, X.; et al. Genome sequencing and comparative genomics of honey bee microsporidia, Nosema apis reveal novel insights into host-parasite interactions. BMC Genom. 2013, 14, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornman, R.S.; Chen, Y.P.; Schatz, M.C.; Street, C.; Zhao, Y.; Desany, B.; Egholm, M.; Hutchison, S.; Pettis, J.S.; Lipkin, W.I.; et al. Genomic Analyses of the Microsporidian Nosema ceranae, an Emergent Pathogen of Honey Bees. PLoS Pathog. 2009, 5, e1000466. [Google Scholar] [CrossRef]
- Cornman, S.R.; Schatz, M.C.; Johnston, S.J.; Chen, Y.P.; Pettis, J.; Hunt, G.; Bourgeois, L.; Elsik, C.; Anderson, D.; Grozinger, C.M.; et al. Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genom. 2010, 11, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigot, D.; Dalmon, A.; Roy, B.; Hou, C.; Germain, M.; Romary, M.; Deng, S.; Diao, Q.; Weinert, L.A.; Cook, J.M.; et al. The discovery of Halictivirus resolves the Sinaivirus phylogeny. J. Gen. Virol. 2017, 98, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.A.-O. Relative Abundance and Molecular Evolution of Lake Sinai Virus (Sinaivirus) Clades. PeerJ 2019, 7, e6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanowicz, D.D.; Wu-Smart, J.Y.; Olgun, T.; Smart, A.H.; Otto, C.R.V.; Lopez, D.; Evans, J.D.; Cornman, R. An updated genetic marker for detection of Lake Sinai Virus and metagenetic applications. PeerJ 2020, 8, e9424. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Moustafa, I.M.; Korboukh, V.K.; Arnold, J.J.; Smidansky, E.D.; Marcotte, L.L.; Gohara, D.W.; Yang, X.; Sánchez-Farrán, M.A.; Filman, D.; Maranas, J.K.; et al. Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase. J. Biol. Chem. 2014, 289, 36229–36248. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S. Within-Host Viral Diversity: A Window into Viral Evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Thaduri, S.; Locke, B.; Granberg, F.; de Miranda, J.R. Temporal changes in the viromes of Swedish Varroa-resistant and Varroa-susceptible honeybee populations. PLoS ONE 2018, 13, e0206938. [Google Scholar] [CrossRef]
- Yue, C. RT-PCR analysis of Deformed wing virus in honeybees (Apis mellifera) and mites (Varroa destructor). J. Gen. Virol. 2005, 86, 3419–3424. [Google Scholar] [CrossRef] [PubMed]
- McNeil, D.J.; McCormick, E.; Heimann, A.C.; Kammerer, M.; Douglas, M.R.; Goslee, S.C.; Grozinger, C.M.; Hines, H.M. Bumble bees in landscapes with abundant floral resources have lower pathogen loads. Sci. Rep. 2020, 10, 22306. [Google Scholar] [CrossRef]
- Annoscia, D.; Brown, S.P.; Di Prisco, G.; De Paoli, E.; Del Fabbro, S.; Frizzera, D.; Zanni, V.; Galbraith, D.A.; Caprio, E.; Grozinger, C.M.; et al. Haemolymph removal by Varroa mite destabilizes the dynamical interaction between immune effectors and virus in bees, as predicted by Volterra’s model. Proc. Biol. Sci. 2019, 286, 20190331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Heerman, M.; Peng, W.; Evans, J.D.; Rose, R.; De Grandi-Hoffman, G.; Simone-Finstrom, M.; Li, J.; Li, Z.; Cook, S.C.; et al. The Dynamics of Deformed Wing Virus Concentration and Host Defensive Gene Expression after Varroa Mite Parasitism in Honey Bees, Apis mellifera. Insects 2019, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zioni, N.; Soroker, V.; Chejanovsky, N. Replication of Varroa destructor virus 1 (VDV-1) and a Varroa destructor virus 1–deformed wing virus recombinant (VDV-1–DWV) in the head of the honey bee. Virology 2011, 417, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Arévalo, S.; Fernández-Carrión, E.; Goyache, J.; Molero, F.; Puerta, F.; Sánchez-Vizcaíno, J.M. High Load of Deformed Wing Virus and Varroa destructor Infestation Are Related to Weakness of Honey Bee Colonies in Southern Spain. Front. Microbiol. 2019, 10, 1331. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef]
- Genersch, E. Honey bee pathology: Current threats to honey bees and beekeeping. Appl. Microbiol. Biotechnol. 2010, 87, 87–97. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, A.J.; Genersch, E. Honey bee colony losses and associated viruses. Curr. Opin. Insect Sci. 2015, 8, 121–129. [Google Scholar] [CrossRef]
- Kevill, J.L.; Highfield, A.; Mordecai, G.J.; Martin, S.J.; Schroeder, D.C. ABC Assay: Method Development and Application to Quantify the Role of Three DWV Master Variants in Overwinter Colony Losses of European Honey Bees. Viruses 2017, 9, 314. [Google Scholar] [CrossRef] [Green Version]
- Dalmon, A.; Desbiez, C.; Coulon, M.; Thomasson, M.; Le Conte, Y.; Alaux, C.; Vallon, J.; Moury, B. Evidence for positive selection and recombination hotspots in Deformed wing virus (DWV). Sci. Rep. 2017, 7, 41045. [Google Scholar] [CrossRef] [PubMed]
- Dubois, E.; Dardouri, M.; Schurr, F.; Cougoule, N.; Sircoulomb, F.; Thiéry, R. Outcomes of honeybee pupae inoculated with deformed wing virus genotypes A and B. Apidologie 2020, 51, 18–34. [Google Scholar] [CrossRef] [Green Version]
- Mockel, N.; Gisder, S.; Genersch, E. Horizontal transmission of deformed wing virus: Pathological consequences in adult bees (Apis mellifera) depend on the transmission route. J. Gen. Virol. 2011, 92, 370–377. [Google Scholar] [CrossRef]
- Ravoet, J.; De Smet, L.; Wenseleers, T.; de Graaf, D.C. Genome sequence heterogeneity of Lake Sinai Virus found in honey bees and Orf1/RdRP-based polymorphisms in a single host. Virus Res. 2015, 201, 67–72. [Google Scholar] [CrossRef]
- Chevin, A.; Coutard, B.; Blanchard, P.; Dabert-Gay, A.-S.; Ribière-Chabert, M.; Thiéry, R. Characterisation of Structural Proteins from Chronic Bee Paralysis Virus (CBPV) Using Mass Spectrometry. Viruses 2015, 7, 3329–3344. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Pascall, D.J.; Tinsley, M.C.; Obbard, D.J.; Wilfert, L. Host evolutionary history predicts virus prevalence across bumblebee species. bioRxiv 2019. [Google Scholar] [CrossRef]
- Carrillo-Tripp, J.; Dolezal, A.G.; Goblirsch, M.J.; Miller, W.A.; Toth, A.L.; Bonning, B.C. In vivo and in vitro infection dynamics of honey bee viruses. Sci. Rep 2016, 6, S50. [Google Scholar] [CrossRef]
- Guo, Y.; Goodman, C.L.; Stanley, D.W.; Bonning, B.C. Cell Lines for Honey Bee Virus Research. Viruses 2020, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Goblirsch, M.J.; Spivak, M.S.; Kurtti, T.J. A Cell Line Resource Derived from Honey Bee (Apis mellifera) Embryonic Tissues. PLoS ONE 2013, 8, e69831. [Google Scholar] [CrossRef] [Green Version]
- McMahon, D.P.; Fürst, M.A.; Caspar, J.; Theodorou, P.; Brown, M.J.F.; Paxton, R.J. A sting in the spit: Widespread cross-infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 2015, 84, 615–624. [Google Scholar] [CrossRef]
- Graystock, P.; Ng, W.H.; Parks, K.; Tripodi, A.D.; Muñiz, P.A.; Fersch, A.A.; Myers, C.R.; McFrederick, Q.S.; McArt, S.H. Dominant bee species and floral abundance drive parasite temporal dynamics in plant-pollinator communities. Nat. Ecol. Evol. 2020, 4, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Ostfeld, R.S.; Keesing, F. Biodiversity and Disease Risk: The Case of Lyme Disease. Conserv. Biol. 2000, 14, 722–728. [Google Scholar] [CrossRef]
- Figueroa, L.L.; Grab, H.; Ng, W.H.; Myers, C.R.; Graystock, P.; McFrederick, Q.S.; McArt, S.H. Landscape simplification shapes pathogen prevalence in plant-pollinator networks. Ecol. Lett. 2020, 23, 1212–1222. [Google Scholar] [CrossRef]
- Luis, A.D.; Kuenzi, A.J.; Mills, J.N. Species diversity concurrently dilutes and amplifies transmission in a zoonotic host-pathogen system through competing mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 7979–7984. [Google Scholar] [CrossRef] [Green Version]
- De Grandi-Hoffman, G.; Chen, Y. Nutrition, immunity and viral infections in honey bees. Curr. Opin. Insect Sci. 2015, 10, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Alaux, C.; Dantec, C.; Parrinello, H.; Le Conte, Y. Nutrigenomics in honey bees: Digital gene expression analysis of pollen’s nutritive effects on healthy and varroa-parasitized bees. BMC Genom. 2011, 12, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolezal, A.G.; Toth, A.L. Feedbacks between nutrition and disease in honey bee health. Curr. Opin. Insect Sci. 2018, 26, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Wenner, A.M.; Meade, D.E.; Friesen, L.J. Recruitment, Search Behavior, and Flight Ranges of Honey Bees1. Am. Zool 1991, 31, 768–782. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daughenbaugh, K.F.; Kahnonitch, I.; Carey, C.C.; McMenamin, A.J.; Wiegand, T.; Erez, T.; Arkin, N.; Ross, B.; Wiedenheft, B.; Sadeh, A.; et al. Metatranscriptome Analysis of Sympatric Bee Species Identifies Bee Virus Variants and a New Virus, Andrena-Associated Bee Virus-1. Viruses 2021, 13, 291. https://doi.org/10.3390/v13020291
Daughenbaugh KF, Kahnonitch I, Carey CC, McMenamin AJ, Wiegand T, Erez T, Arkin N, Ross B, Wiedenheft B, Sadeh A, et al. Metatranscriptome Analysis of Sympatric Bee Species Identifies Bee Virus Variants and a New Virus, Andrena-Associated Bee Virus-1. Viruses. 2021; 13(2):291. https://doi.org/10.3390/v13020291
Chicago/Turabian StyleDaughenbaugh, Katie F., Idan Kahnonitch, Charles C. Carey, Alexander J. McMenamin, Tanner Wiegand, Tal Erez, Naama Arkin, Brian Ross, Blake Wiedenheft, Asaf Sadeh, and et al. 2021. "Metatranscriptome Analysis of Sympatric Bee Species Identifies Bee Virus Variants and a New Virus, Andrena-Associated Bee Virus-1" Viruses 13, no. 2: 291. https://doi.org/10.3390/v13020291