
Reanalysis of Trio Whole-Genome Sequencing Data Doubles the Yield in Autism Spectrum Disorder: De Novo Variants Present in Half



Abstract
:1. Introduction
2. Results
2.1. Subject Characteristics
2.2. De Novo Variants
2.3. Inherited Variants
2.4. Combined Primary Diagnostic Variants and Yield from Laboratory Report
2.5. Genotype–Phenotype Correlation
2.6. Candidate Polygenic Modifier Variants
2.7. Actionability of Genetic Results
3. Discussion
3.1. Our Subjects Represent the Broad Phenotype of Autism in Terms of Sex, Severity, and Co-Morbidities
3.2. WGS with Comprehensive Sequence Reanalysis Revealed High Sensitivity for Identification of Primary Diagnostic Variants (PDVs) in Our Autism Subjects
3.3. Autism as a Polygenic/Multifactorial Condition
3.4. Variant Curation Comparison to ACMG Guidelines
3.5. Limitations of the Study
3.6. Risks and Additional Costs
3.7. Implications of Our Data to a Greater Understanding of ASD
4. Subjects and Methods
4.1. Subjects
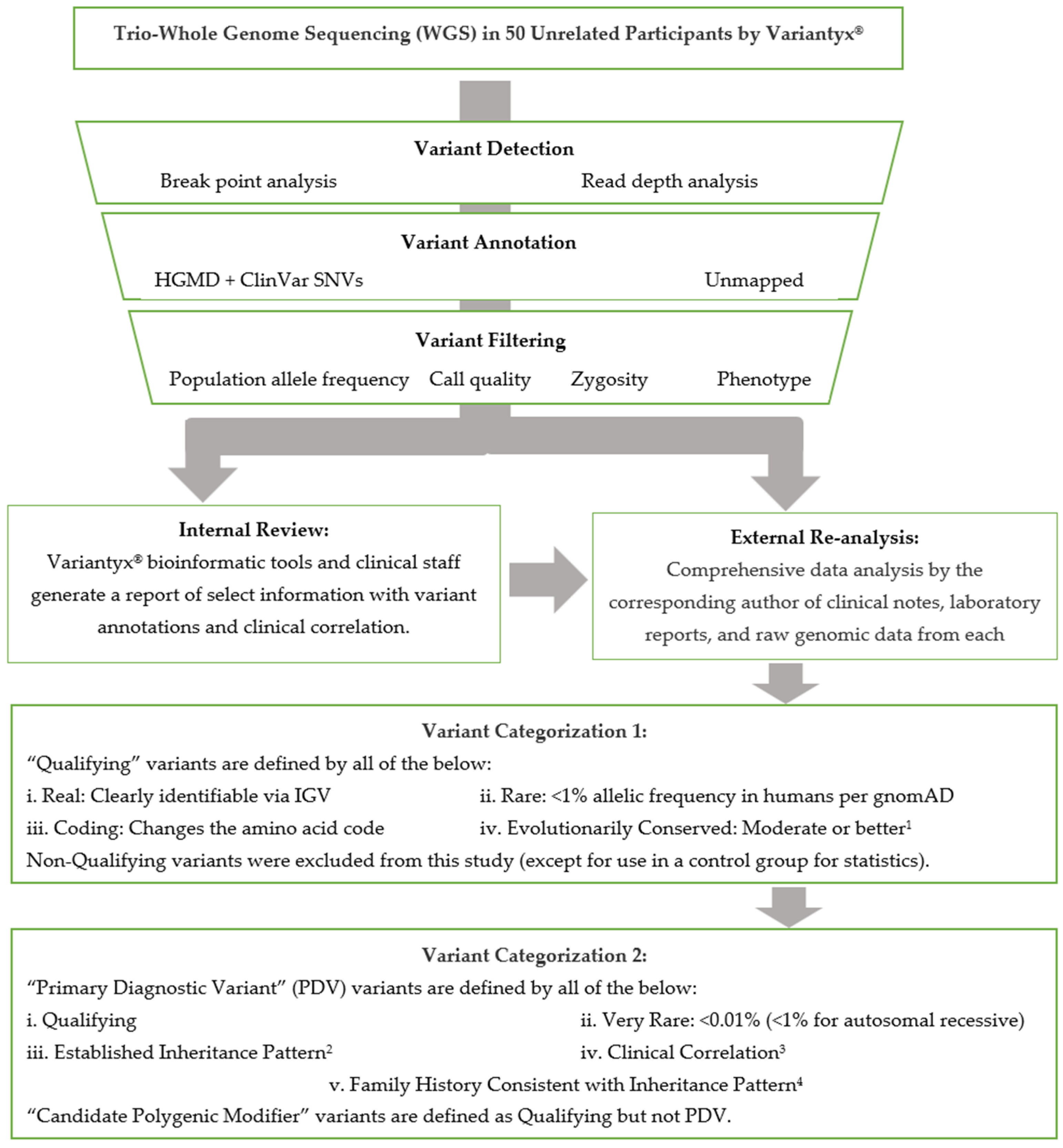
4.2. Sequencing and Data Analysis
4.3. Gene Categorization
4.4. Variant Categorization
- De novo: Any very rare (<1/10,000), de novo, Qualifying variant in a Direct gene, with clinical correlation. Single-copy variants in genes with well-established autosomal recessive inheritance were excluded.
- X-linked: Any very rare (<1/10,000), inherited, hemizygous, Qualifying variant in a Direct gene on the X-chromosome, with clinical correlation.
- Autosomal recessive: Any rare (<1/100), inherited homozygous or in trans compound heterozygous Qualifying variants in a Direct gene on an autosome, with clinical correlation.
- Autosomal dominant: Any very rare (<1/10,000), inherited Qualifying variant in a Direct gene on an autosome, with clinical correlation, and with the parent harboring that variant being affected with significant neurodevelopmental disease. “Significant” was defined as substantially affecting their quality-of-life per the family and in the judgment of the corresponding author.
- Maternal inheritance: Any very rare (<1/10,000), Qualifying variant in a mitochondrial-encoded gene (mtDNA) with clinical correlation that is either heteroplasmic (with the minor allele present at 40–98%) and/or with a pedigree highly suggestive of maternal inheritance.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; et al. Environmental risk factors and biomarkers for autism spectrum disorder: An umbrella review of the evidence. Lancet Psychiatry 2019, 6, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.T.; Friedman, E.; Jenkins, E.C.; Brooks, J.; Wisniewski, K.; Raguthu, S.; French, J.H. Association of fragile X syndrome with autism. Lancet 1982, 319, 100. [Google Scholar] [CrossRef]
- Steffenburg, S.; Gillberg, C.; Hellgren, L.; Andersson, L.; Gillberg, I.C.; Jakobsson, G.; Bohman, M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry 1989, 30, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- Dietert, R.R.; Dietert, J.M.; DeWitt, J.C. Environmental risk factors for autism. Emerg. Health Threat. J. 2011, 4, 7111. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Walker, M.F.; Carriero, N.J.; DiCola, M.; Willsey, A.J.; Adam, Y.Y.; Waqar, Z.; Gonzalez, L.E.; Overton, J.D.; Frahm, S.; et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 2014, 9, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Iossifov, I.; O’roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’Ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 2015, 18, 191–198. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef]
- SFARI Gene. Available online: https://gene.sfari.org (accessed on 20 June 2023).
- AutDB. Available online: http://www.mindspec.org/autdb.html (accessed on 25 May 2023).
- Kreiman, B.L.; Boles, R.G. State of the art of genetic testing for patients with autism: A practical guide for clinicians. Semin. Pediatr. Neurol. 2020, 34, 100804. [Google Scholar] [CrossRef]
- Shevell, M.I.; Majnemer, A.; Rosenbaum, P.; Abrahamowicz, M. Etiologic yield of autistic spectrum disorders: A prospective study. J. Child Neurol. 2001, 16, 509–512. [Google Scholar] [CrossRef]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Mol. Autism 2019, 10, 33. [Google Scholar] [CrossRef]
- Schaefer, G.B.; Lutz, R.E. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet. Med. 2006, 8, 549–556. [Google Scholar] [CrossRef]
- Jacquemont, M.L.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Le Merrer, M.; Heron, D.; De Blois, M.C.; et al. Array-based comparative genomic hybridization identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 2006, 43, 843–849. [Google Scholar] [CrossRef]
- Leppa, V.M.; Kravitz, S.N.; Martin, C.L.; Andrieux, J.; Le Caignec, C.; Martin-Coignard, D.; DyBuncio, C.; Sanders, S.J.; Lowe, J.K.; Cantor, R.M.; et al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet. 2016, 99, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Du, X.; Gao, X.; Liu, X.; Shen, L.; Wang, K.; Fan, Y.; Sun, Y.; Luo, X.; Liu, H.; Wang, L.; et al. Genetic diagnostic evaluation of trio-based whole exome sequencing among children with diagnosed or suspected autism spectrum disorder. Front. Genet. 2018, 9, 594. [Google Scholar] [CrossRef]
- Miyake, N.; Tsurusaki, Y.; Fukai, R.; Kushima, I.; Okamoto, N.; Ohashi, K.; Nakamura, K.; Hashimoto, R.; Hiraki, Y.; Son, S.; et al. Molecular diagnosis of 405 individuals with autism spectrum disorder. Eur. J. Hum. Genet. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Yuen, R.K.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Ju, J.; Mei, J.; Shi, Y.; He, M.; et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013, 93, 249–263. [Google Scholar] [CrossRef]
- Abdi, M.; Aliyev, E.; Trost, B.; Kohailan, M.; Aamer, W.; Syed, N.; Shaath, R.; Devadoss Gandhi, G.; Engchuan, W.; Howe, J.; et al. Genomic architecture of autism spectrum disorder in Qatar: The BARAKA-Qatar Study. Genome Med. 2023, 15, 81. [Google Scholar] [CrossRef]
- Sheth, F.; Shah, J.; Jain, D.; Shah, S.; Patel, H.; Patel, K.; Solanki, D.I.; Iyer, A.S.; Menghani, B.; Mhatre, P.; et al. Comparative yield of molecular diagnostic algorithms for autism spectrum disorder diagnosis in India: Evidence supporting whole exome sequencing as first tier test. BMC Neurol. 2023, 23, 292. [Google Scholar] [CrossRef]
- Higashimoto, T.; Baldwin, E.E.; Gold, J.I.; Boles, R.G. Reflex sympathetic dystrophy: Complex regional pain syndrome type I in children with mitochondrial disease and maternal inheritance. Arch. Dis. Child. 2008, 93, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Warren, Z.; Robinson Williams, A.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Shaw, K.A. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Autism Speaks. Available online: https://www.autismspeaks.org/autism-statistics-asd (accessed on 23 July 2023).
- Gundogdu, B.S.; Gaitanis, J.; Adams, J.B.; Rossignol, D.A.; Frye, R.E. Age-Related Changes in Epilepsy Characteristics and Response to Antiepileptic Treatment in Autism Spectrum Disorders. J. Pers. Med. 2023, 13, 1167. [Google Scholar] [CrossRef]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Michaelson, J.J.; Shi, Y.; Gujral, M.; Zheng, H.; Malhotra, D.; Jin, X.; Jian, M.; Liu, G.; Greer, D.; Bhandari, A.; et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 2012, 151, 1431–1442. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, B.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.L.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef]
- Kristmundsdottir, S.; Jonsson, H.; Hardarson, M.T.; Palsson, G.; Beyter, D.; Eggertsson, H.P.; Gylfason, A.; Sveinbjornsson, G.; Holley, G.; Stefansson, O.A.; et al. Sequence variants affecting the genome-wide rate of germline microsatellite mutations. Nat. Commun. 2023, 14, 3855. [Google Scholar] [CrossRef]
- Felipič, M. Mechanisms of cadmium induced genomic instability. Mutat. Res. 2011, 733, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Xu, J.; Lin, Y.L.; Cabrera, R.M.; Chen, Q.; Zhang, C.; Steele, J.W.; Han, X.; Gross, S.S.; Wlodarczyk, B.J.; et al. Excess folic acid intake increases DNA de novo point mutations. Cell Discov. 2023, 9, 22. [Google Scholar] [CrossRef]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr. Environ. Health Rep. 2022, 9, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Integrative Genomics Viewer. Available online: https://software.broadinstitute.org/software/igv/ (accessed on 21 June 2023).
- Neerman, N.; Faust, G.; Meeks, N.; Modai, S.; Kalfon, L.; Falik-Zaccai, T.; Kaplun, A. A clinically validated whole genome pipeline for structural variant detection and analysis. BMC Genom. 2019, 20 (Suppl. 8), 545. [Google Scholar] [CrossRef] [PubMed]
- VariCarta. Available online: https://varicarta.msl.ubc.ca/index (accessed on 20 June 2023).
- Wang, T.; Kim, C.N.; Bakken, T.E.; Gillentine, M.A.; Henning, B.; Mao, Y.; Gilissen, C.; SPARKConsortium Nowakowski, T.J.; Eichler, E.E. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proc. Natl. Acad. Sci. USA 2022, 119, e2203491119. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Almeida, T.F.D. Molecular Diagnosis of Autism Spectrum Disorder through Whole Exome Sequencing. Ph.D. Dissertation, Universidade de São Paulo, São Paulo, Brazil, 2018. [Google Scholar]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Webber, C. The roles of FMRP-regulated genes in autism spectrum disorder: Single-and multiple-hit genetic etiologies. Am. J. Hum. Genet. 2013, 93, 825–839. [Google Scholar] [CrossRef]
- Uddin, M.; Tammimies, K.; Pellecchia, G.; Alipanahi, B.; Hu, P.; Wang, Z.; Pinto, D.; Lau, L.; Nalpathamkalam, T.; Marshall, C.R.; et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet. 2014, 46, 742–747. [Google Scholar] [CrossRef]
- Bar, O.; Ebenau, L.; Weiner, K.; Mintz, M.; Boles, R.G. Whole exome/genome sequencing in cyclic vomiting syndrome reveals multiple candidate genes, suggesting a model of elevated intracellular cations and mitochondrial dysfunction. Front. Neurol. 2023, 14, 1151835. [Google Scholar] [CrossRef]
- University of California Santa Cruz Genomic Institute UCSC Genome Browser. Available online: https://genome.ucsc.edu/ (accessed on 21 June 2023).
- Rai, V. Association of methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism with autism: Evidence of genetic susceptibility. Metab. Brain Dis. 2016, 31, 727–735. [Google Scholar] [CrossRef] [PubMed]
- MITOMAP A Human Mitochondrial Database. Available online: www.mitomap.org/MITOMAP (accessed on 20 July 2023).
- GraphPad by Dotmatics. Available online: https://www.graphpad.com/quickcalcs/contingency1.cfm (accessed on 21 June 2023).
- MedCalc®. Available online: https://www.medcalc.org/calc/odds_ratio.php (accessed on 21 June 2023).


{kind=link}
| Subject # | Age and Sex | Developmental | Verbal 1 | Seizure/EEG | Regression | Other Neuropsychiatric 2 | Other Phenotypes 3 |
|---|---|---|---|---|---|---|---|
| 1 | 20M | severe ID | no | no/spike-wave | no, plateau | ||
| 2 | 10F | moderate ID | reduced | seizures | multiple | OCD, tics, anxiety | fatigue, pain, GI |
| 3 | 14F | moderate ID | reduced | none | multiple | OCD, anxiety | GI |
| 4 | 5M | moderate ID | reduced | none | no | anxiety | GI |
| 5 | 22M | multiple LDs | yes | none | no | anxiety | |
| 6 | 10M | severe ID (mild in sister) | no (yes in sister) | none (none) | one (one) | GI | |
| 7 | 13M | multiple LDs | yes | none | no | ||
| 8 | 6F | mild–moderate ID | reduced | none | one | ||
| 9 | 22M | severe ID | no | myoclonic | no | ||
| 10 | 5M | likely normal | very little | none | no | ||
| 11 | 12M | mild ID | yes | none | yes, chronic | choreiform, weakness, ataxia | systemic inflammatory response syndrome, immunodeficiency, autonomic instability, GI/parenteral nutrition, ventilation dependence |
| 12 | 4F | severe ID | no | seizures | no | ||
| 13 | 26M | severe ID | NR | none | no | ataxia, choreoathetosis, cerebral palsy | |
| 14 | 14M | multiple LDs | yes | none | episodic | OCD, tics | fevers to 40.5 C |
| 15 | 19M | mild–moderate ID | NR | seizures | episodic | tics, psychosis/cyclical catatonia | |
| 16 | 6M | multiple LDs | yes | none | one | ||
| 17 | 10M | severe ID | no | none | no | ||
| 18 | 12F | multiple LDs | yes | none | no | tics | |
| 19 | 14M | moderate ID | no | none | multiple | tics | CVID |
| 20 | 14M | normal | no | seizures | one | tics | |
| 21 | 16M | moderate ID | yes | seizures | one | OCD, anxiety, depression | pain, cyclic vomiting |
| 22 | 6M | severe ID | no | none | multiple | ||
| 23 | 5M | moderate ID | no | none | one | OCD, Brown syndrome | |
| 24 | 25F | mild–moderate ID | yes | seizures | no | OCD, anxiety, psychosis | GI |
| 25 | 16F | mild ID | yes | none | no | OCD, anxiety | fatigue |
| 26 | 5M | moderate ID | yes | none | likely | fatigue, GI | |
| 27 | 16M | moderate ID | reduced | none | one | OCD, ADD | |
| 28 | 22M | multiple LDs | yes | seizures | no | OCD, anxiety | |
| 29 | 10M | moderate ID | yes | none | one | tics, ADHD | GI |
| 30 | 13M | moderate–severe ID | yes | none | no | GI | |
| 31 | 3M | mild ID | yes | none | one | GI | |
| 32 | 12M | mild ID/LD | yes | none | no | OCD, ADHD, bipolar, POTS, PTSD | GI |
| 33 | 7M | mild ID | yes | none | no | ADD, motor dyspraxia | GI |
| 34 | 19M | severe ID | reduced | none | one | OCD, tics, ADHD | GI |
| 35 | 22M | moderate ID | yes | seizures | no | ADHD | Noonan syndrome |
| 36 | 8F | normal | yes | none | no | OCD, ADHD | fatigue |
| 37 | 5M | mild–moderate ID | no | none | one | GI | |
| 38 | 17M | mild ID | reduced | none | likely | tics | hypogammaglobulinemia/CVID-like |
| 39 | 12M | mild ID | yes | seizures | no | tics, ADHD | pain, GI |
| 40 | 9M | mild LD | yes | none | no | OCD, tics, ADHD, anxiety | hearing loss, GI |
| 41 | 10M | moderate ID/LD | yes | seizures | one | macrosomia | |
| 42 | 13M | mild ID | reduced | neonatal only | multiple | fatigue | |
| 43 | 18F | moderate ID | yes | seizures | no | OCD, ADHD, bipolar, POTS, PTSD | fatigue, pain, GI |
| 44 | 6M | severe ID | reduced | none | multiple | anxiety | |
| 45 | 15M | mild–moderate ID | yes | seizures | no | ADHD | GI |
| 46 | 16F | severe ID | no | seizures | multiple | OCD, anxiety, depression | Turner syndrome, GI, hypogammaglobulinemia |
| 47 | 7M | severe ID | no | none | multiple | tics | |
| 48 | 13F | mild LD | reduced | seizures | no | insomnia | GI, pain |
| 49 | 7M | severe ID | yes | none | one | OCD, tics | GI |
| 50 | 7F | likely normal | reduced | none | likely |
| Participant # | Family History | Inheritance Pattern of Primary Diagnostic Variants | Autism Genes with Qualifying De Novo Variants | Autism Genes with Qualifying Inherited Variants | Lab Identified 1 |
|---|---|---|---|---|---|
| 1 | essentially negative | de novo | EHMT1 | RYR2 | yes |
| 2 | essentially negative | autosomal recessive, CompHet in trans | IVD (AR-CompHet) | yes | |
| 3 | essentially negative | de novo | PGAM5 | CDH15 | no |
| 4 | essentially negative | de novo | COL4A1 | TRAP1 | no |
| 5 | essentially negative | de novo | TRPM2 | no | |
| 6 | sister autism; siblings and mother ADHD | X-linked, mother carrier | THOC2 (XL-Mat), dup with CHL1, RIC1 (AR-CompHet) | no | |
| 7 | brother LD | de novo | del 5 SFARI genes | yes | |
| 8 | essentially negative | de novo | MTMR4 | 2p16.1p16.1 dup with FANCL, 9p13.3p13.3 dup | no |
| 9 | essentially negative | de novo | del with UBE3A | FRMPD4 (XL) | yes |
| 10 | slow speech in twin sister, now normal | N/A | |||
| 11 | affected sister; parents with small fiber neuropathy | de novo | USP20 | CLPX, POLRMT, RELN, SCN10A | no |
| 12 | essentially negative | de novo | SLC41A2 | FLNA (XL) | no |
| 13 | brother LD, ADHD, possible autism | autosomal recessive, homozygous | SLC1A4 (AR-Hom) | yes | |
| 14 | essentially negative | SCN9A, CPT2 | N/A | ||
| 15 | essentially negative | de novo | KCNB1 | MT-TW | yes |
| 16 | essentially negative | X-linked, mother carrier | USP9X (XL-Mat), CIC | no | |
| 17 | essentially negative | de novo | GRIK1 | no | |
| 18 | brother LD; father possible autism | GABRA1, tetrasomy at 14q32q33 with 26 genes | N/A | ||
| 19 | essentially negative | CACNA1A, RIMS1 | N/A | ||
| 20 | essentially negative | N/A | |||
| 21 | sister ADHD | de novo | OCM | SCN10A | no |
| 22 | sister ADHD; mother ADD | X-linked, mother carrier | NLGN4X (XL-Mat), 4p16.1p16.1 dup with SORCS3, 9p24.3p24.3 dup with DOCK8, MT-ND3 | yes | |
| 23 | essentially negative | POLA1 (XL), PAH (AR-CompHet), 22q11.21 11.5-kb del, 14q32.33 27-kb dup, MT-CYB | N/A | ||
| 24 | essentially negative | 7p22.3p22.3x1, DYNC1H1 | N/A | ||
| 25 | essentially negative | de novo | ANKFN1 | SCN2A, ASH1L | no |
| 26 | essentially negative | de novo | CEP170, NUP210 | no | |
| 27 | essentially negative | SCN9A, RYR2 | N/A | ||
| 28 | essentially negative | de novo | ANKRD11 | TRAP1 | yes |
| 29 | essentially negative | N/A | |||
| 30 | brother OCD, autism | de novo | RIF1, AGO3 | PRKCA | no |
| 31 | essentially negative | N/A | |||
| 32 | affected brother; father probable LD | de novo | Large del with GGNBP2 | SETD1B, DEAF1 | yes |
| 33 | only extended relatives affected | autosomal recessive, homozygous | EIF3F (AR-Hom), SCN10A | yes | |
| 34 | father ADD | autosomal dominant, paternal | TCF20 (AD-Pat), MT-TC | no | |
| 35 | essentially negative | de novo | KRAS | MTHFR, HSPG2 | yes |
| 36 | mother and dizygotic twin with autism | autosomal dominant, maternal | RERE (AD-Mat) | yes | |
| 37 | essentially negative | GBE1 (AR-CompHet) | N/A | ||
| 38 | essentially negative | MTHFR, TSC2, MT-CYB | N/A | ||
| 39 | father possible autism | de novo | YTHDF1 | CUX2, 148-kb del with IMMP2L | no |
| 40 | essentially negative | de novo | GRB10, STAT1 | RYR2 | no |
| 41 | essentially negative | de novo | GOLGB1 | no | |
| 42 | essentially negative | de novo | GABRA1 | 16q23.1q23.1, 736.50-kb with ADAMTS18 | yes |
| 43 | essentially negative | de novo | SP8 | MT-CYB | no |
| 44 | mother and sister ADHD | N/A | |||
| 45 | essentially negative | SHROOM4 (XL) | N/A | ||
| 46 | brother ADHD | KMT2E, RYR2, MT-CO3 | N/A | ||
| 47 | essentially negative | SCN4A, KCND2, 7q31.31q31.31x1 del with IGHG2 | N/A | ||
| 48 | sister ADHD, ID, seizures, MELAS | de novo + mtDNA | SPEN | MT-CO1 | yes |
| 49 | brother autism | autosomal recessive, CompHet in trans | Large del with 53% PRODH | ZNF292 (AR-ComHet), Xq22.3q22.3x1, 54 base pairs; includes TBC1D8B | no |
| 50 | essentially negative | de novo | HSPA1A | no |
| Participant # | Gene | Report 1 | Disorder | NDD per HGMD 2 | Protein Function | Cation Trans-port | Redox State | Amino Acids | Ubiquitination | Neurotransmitter | Gene Expression | Cell Division |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| On Lab Report | ||||||||||||
| 1 | EHMT1 | Uncertain | Known | Histone methyltransferase | Yes | |||||||
| 2 | IVD | Candidate POSITIVE | Known | Amino acid metabolism | Yes | |||||||
| 7 | del SFARI x5 3 | POSITIVE | Known | Ubiquitination/cation channel/cholinergic receptor 3 | Yes | Yes | Yes | Yes | ||||
| 9 | UBE3A 3 | POSITIVE | Known | Ubiquitination | Yes | Yes | ||||||
| 13 | SLCIA4 | POSITIVE | Known | Amino acid transport | Yes | |||||||
| 15 | KCNB1 | POSITIVE | Known | Potassium transporter | Yes | |||||||
| 22 | NLGN4X | Likely | Known | Neuronal cell-cell interactions, glutamate receptors | Yes | |||||||
| 28 | ANKRD11 | Likely Positive | Known | Transcription | Yes | |||||||
| 32 | GGNBP2 3 | POSITIVE | Known | Growth suppressor? | ||||||||
| 33 | EIF3F | Uncertain | Known | Translation initiation factor | Yes | |||||||
| 35 | KRAS | Likely Negative | Known | Ras protein, GTPase activity, regulation of cell proliferation | Yes | |||||||
| 36 | RERE | Likely Negative | Known | Transcription | Yes | |||||||
| 42 | GABRA1 | Likely Negative | Known | GABA receptor, chloride channel | Yes | Yes | ||||||
| 48 | SPEN | Likely Positive | Known | Transcription | ||||||||
| 48 (PDV2) | MT-CO1 | Likely Negative | Known | Energy metabolism | Yes | |||||||
| Not on Report | ||||||||||||
| 3 | PGAM5 | Novel | 1 | Programmed cell death, mitophagy | Yes | |||||||
| 4 | COL4A1 | Known | Collagen, structural | |||||||||
| 5 | TRPM2 | Very rare 5 | Calcium channel, oxidative stress | Yes | Yes | |||||||
| 6 | THOC2 | Very rare 6 | Transcription | Yes | ||||||||
| 8 | MTMR4 | Novel | Ubiquitination, vesicular fusion, phagocytosis | Yes | ||||||||
| 11 4 | USP20 | Novel | 2 | Ubiquitination, inflammatory signaling | Yes | |||||||
| 12 | SLC41A2 | Novel | 3 | Cation 2+ transporter, including magnesium | Yes | |||||||
| 16 | USP9X | Known | Ubiquitination, separating sister chromatids, axonal growth | Yes | Yes | |||||||
| 17 | GRIK1 | Novel | 3 | Glutamate ionotropic receptor kainate type | Yes | Yes | ||||||
| 21 | OCM | Novel | 0 | Calcium buffering | Yes | |||||||
| 25 | ANKFN1 | Nove l 7 | Orientation of mitotic spindle and cell polarity | Yes | ||||||||
| 26 | GEP170 | Very rare 8 | Centrosome component | Yes | ||||||||
| 30 | RIFI | Novel | 3 | Cell check point | Yes | |||||||
| 30 (PDV2) | AG03 | Novel | 3 | Transcription | Yes | |||||||
| 34 | TCF20 | Known | Transcription | Yes | ||||||||
| 39 | YTHDFI | Novel | Binds m6A-containg mRNAs | Yes | ||||||||
| 40 | GRBIO | Novel | 5 | Involved in multiple cell signaling cascades | ||||||||
| 41 | GOLGB1 | Novel | 3 | Golgi crosslinking | ||||||||
| 43 | SP8 | Novel | 0 9 | Transcription | Yes | |||||||
| 49 | PRODH | Known | 0 9 | Amino acid metabolism | Yes | |||||||
| 50 | HSPA1A | Novel | Chaperone | Yes |
| Platform | Variant Types Detected | Subjects | Diagnostic Yield | De Novo Yield 1 | Reference |
|---|---|---|---|---|---|
| FMR-1 | FMR-1 | 50 | 2% | Shevell et al., 2001 [21] | |
| FMR-1 | FMR-1 | 502 | 1.3% | Munnich et al., 2019 [22] | |
| Cytogenics | CNV, MECP-2 | 32 | 41% | Schafer and Lutz, 2006 [23] | |
| Microarray | CNVs | 29 | 27.5% | Jacquemont et al., 2006 [24] | |
| Microarray | CNVs | 1532 | 3.0% | Leppa et al., 2016 [25] | |
| Microarray | CNVs | 502 | 8.8% | Munnich et al., 2019 [22] | |
| Targeted Sequencing 2 | SNVs and indels | >11,730 | 5.7% | Stessman et al., 2017 [26] | |
| Trio-WES | SNVs and indels | 17 | ~90% 3 | 18% | Al-Mubarak et al., 2017 [29] |
| Trio-WES | SNVs and indels | 80 simplex families | 9.2% ASD, 6.7% suspected ASD | 8% | Du et al., 2018 [27] |
| Trio-WES | SNVs, small indels, and CNVs | 405 | 16% | 15% | Miyake et al., 2023 [28] |
| Trio-WGS | Essentially all variants | 32 | 50% | 19% | Jiang et al., 2013 [30] |
| Trio-WGS | Essentially all variants | 100 | 41% | 14% | Abdi et al., 2023 [31] |
| Trio-WGS | Essentially all variants | 101 | 33% | 20% | Sheth et al., 2023 [32] |
| Direct Genes: Direct Association with ASD: |
| • A1—Indicating the highest association, it was designated to SFARI [18] 1 (or 1S “Syndromic”) ranking or a 4 -or 5-star AutDB [19] evidence score, whether ranked as such by those websites or by the present authors using their published criteria. • A2—Indicating genes with strong, but not overwhelming, association with ASD, it was designated to SFARI 2 (2S) ranking or with a 3-star AutDB evidence score, per those websites or the present authors using their criteria. • A3—Indicating the weakest level with direct association with ASD, it was designated to SFARI 3 (3S) ranking or with a 2-star AutDB evidence score as per those websites or the present authors. Additionally, some genes were placed in this category by the present authors due to findings of replicated or un-replicated statistical significance in association studies, as reported in ASD with one or more of the following: (i) an exonic de novo variant with >20 Combined Annotation Dependent Depletion (CADD) score [49,50] for genes related to another neurodevelopmental or neuropsychiatric disorder (such as bipolar disorder, schizophrenia, ADHD, and intellectual disability), (ii) a variant identified in a case with ASD [50] in a gene associated with another NDD, (iii) reported in ASD in ≥10 reported copy number variants (CNVs) per AutDB, and/or (iv) an ASD-like phenotype in an animal model. Lastly, some genes that qualified for the B1 category (as described below) became A3 genes if they were intolerant to loss-of-function mutations (supplemental material of [51], also seen in attachment 10 of [52]) and were either Fragile X syndrome genes that were found more enriched in an ASD group than a control group [53,54], also seen in attachment 4 of [52] or occurred in brain-expressed exons that were found with significant accumulation of de novo mutations in individuals with ASD when compared to controls [55] (also seen in attachment 1 of [52]). |
| Non-Direct genes: Indirect or Absent Association to ASD: |
| • B1—Indicating genes with an indirect association with ASD, was designated to (i) genes with a published direct association with any Direct gene, (ii) genes with direct association with another NDD phenotype that is itself associated with ASD (e.g., AD/HD, intellectual disability, schizophrenia, bipolar) with CADD ≥ 20, and/or (iii) genes in pathways in which ASD clearly has been associated. ASD-associated pathways include brain ion-channels, energy metabolism, amino acid metabolism, protein ubiquitination, neuronal cell development, cytoskeleton, epigenetic regulation, inflammation or immunodeficiency, and phosphatidylinositol signaling. • B2—Indicating genes with unknown association with ASD, designated to non-Direct genes neither meeting “B1” nor “B3” criteria. In practice, most “B2” genes occur in genes of uncertain function or in pathways with weak association with ASD. • B3—Indicating genes that are unlikely to be ASD related, it was designated to genes with known effects predominately in non-nervous tissues. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bar, O.; Vahey, E.; Mintz, M.; Frye, R.E.; Boles, R.G. Reanalysis of Trio Whole-Genome Sequencing Data Doubles the Yield in Autism Spectrum Disorder: De Novo Variants Present in Half. Int. J. Mol. Sci. 2024, 25, 1192. https://doi.org/10.3390/ijms25021192
Bar O, Vahey E, Mintz M, Frye RE, Boles RG. Reanalysis of Trio Whole-Genome Sequencing Data Doubles the Yield in Autism Spectrum Disorder: De Novo Variants Present in Half. International Journal of Molecular Sciences. 2024; 25(2):1192. https://doi.org/10.3390/ijms25021192
Chicago/Turabian StyleBar, Omri, Elizabeth Vahey, Mark Mintz, Richard E. Frye, and Richard G. Boles. 2024. "Reanalysis of Trio Whole-Genome Sequencing Data Doubles the Yield in Autism Spectrum Disorder: De Novo Variants Present in Half" International Journal of Molecular Sciences 25, no. 2: 1192. https://doi.org/10.3390/ijms25021192