
PolyQ-Expansion Causes Mitochondria Fragmentation Independent of Huntingtin and Is Distinct from Traumatic Brain Injury (TBI)/Mechanical Stress-Mediated Fragmentation Which Results from Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drosophila Genetics
2.2. Larval Immunostaining and Quantification of Mitochondria Size
2.3. TUNEL Assay
2.4. Mdivi-1 and L-NAME Treatment
2.5. In Vivo Mitochondria Health Reporters
2.6. TMRM Assay
2.7. JC-1 Assay
2.8. Quantification of Nitric Oxide Levels
2.9. Mechanical Stress-TBI by Vortexing
2.10. Western Blot and Analysis
2.11. Statistical Analysis
3. Results
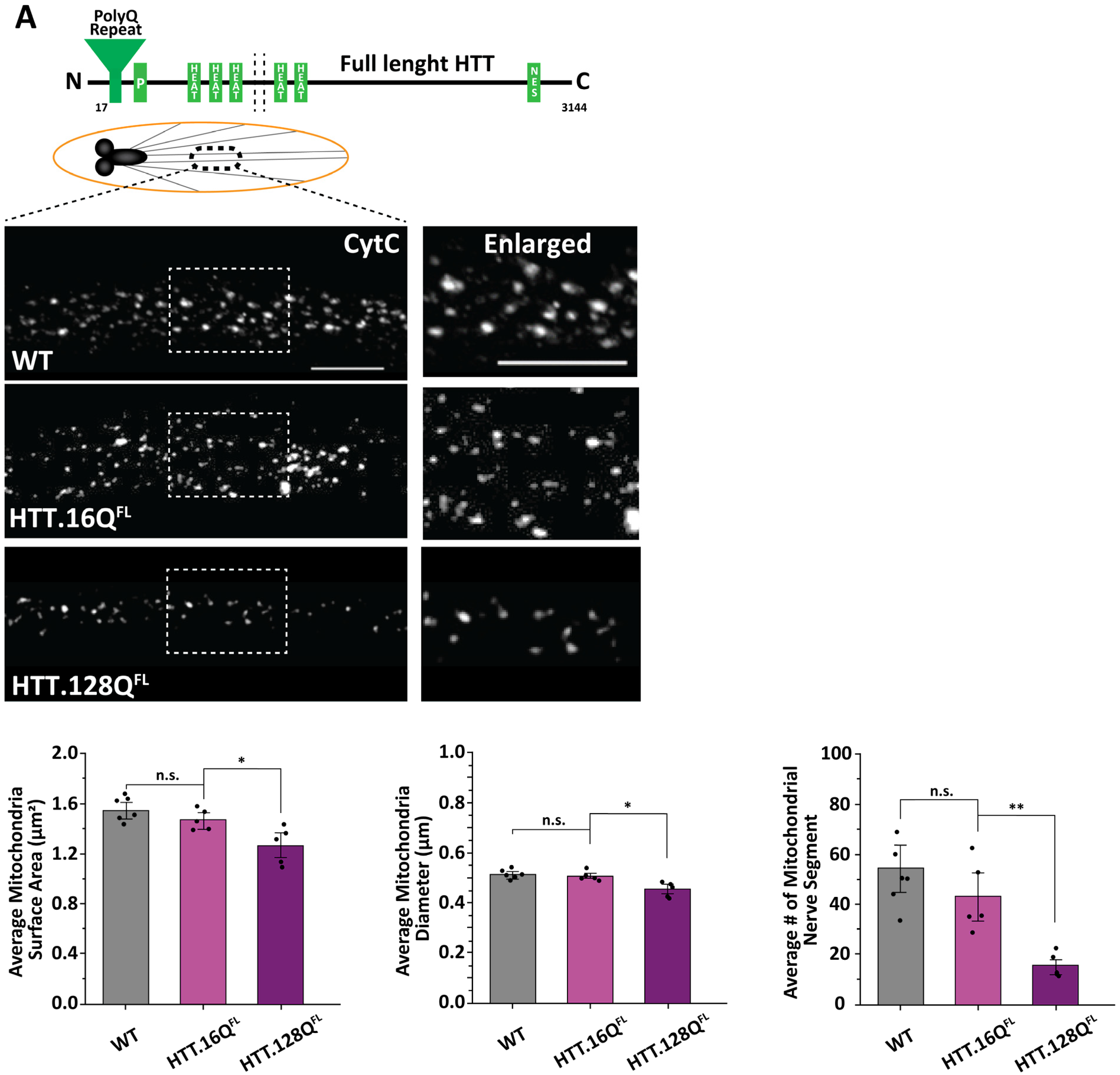
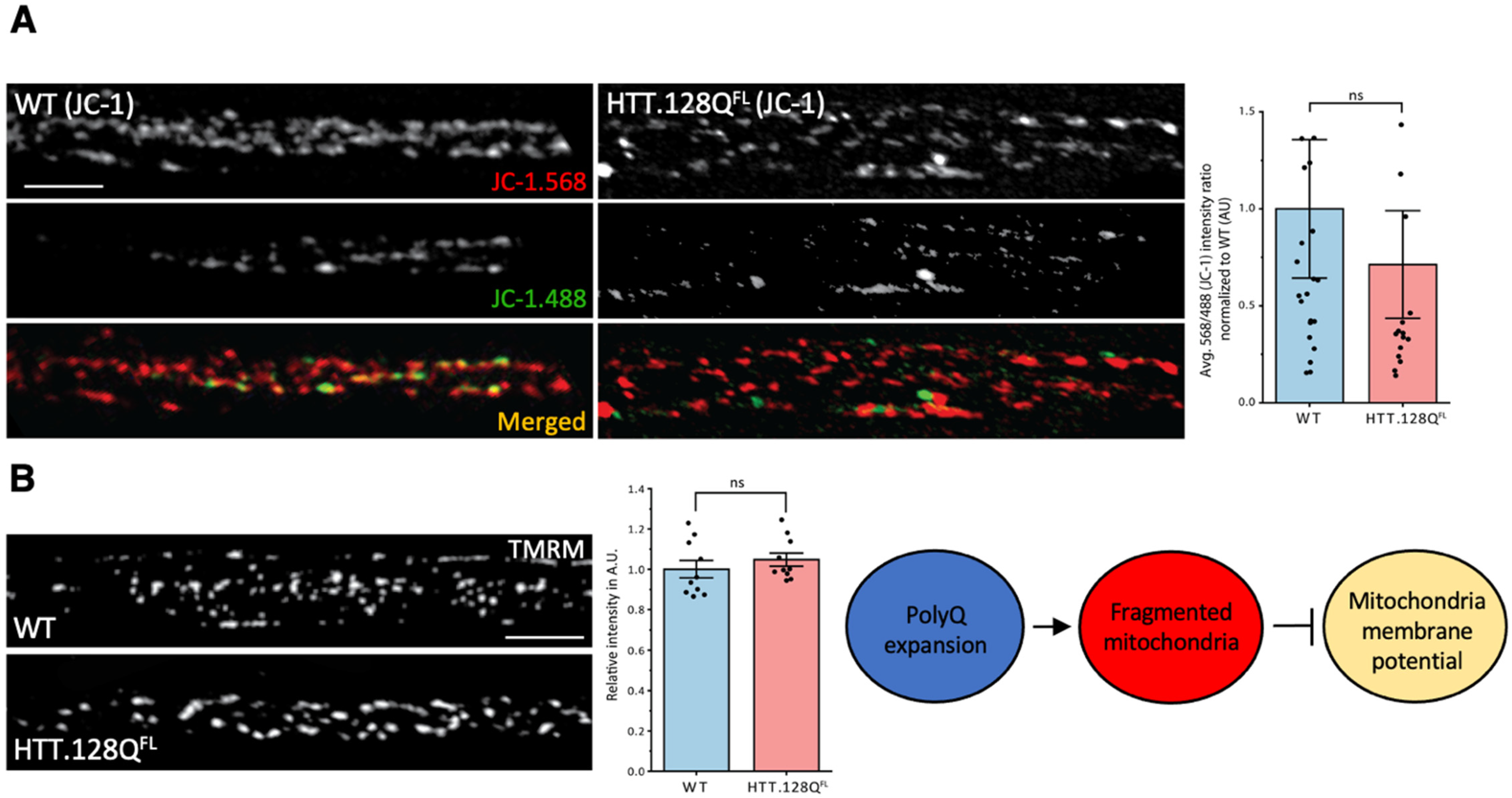
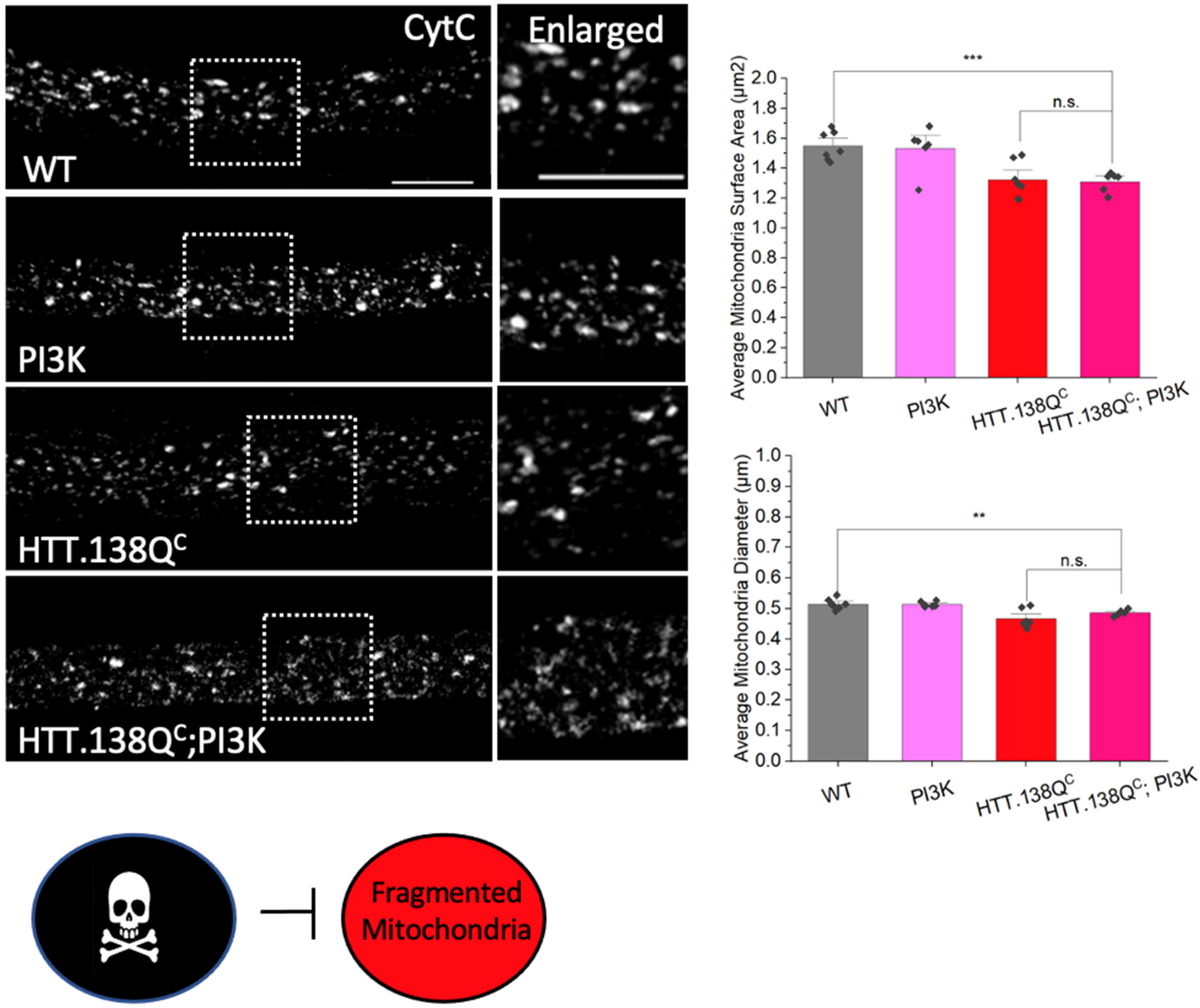
3.1. Excess Pathogenic Huntingtin Causes Mitochondrial Fragmentation in Drosophila Larval Axons
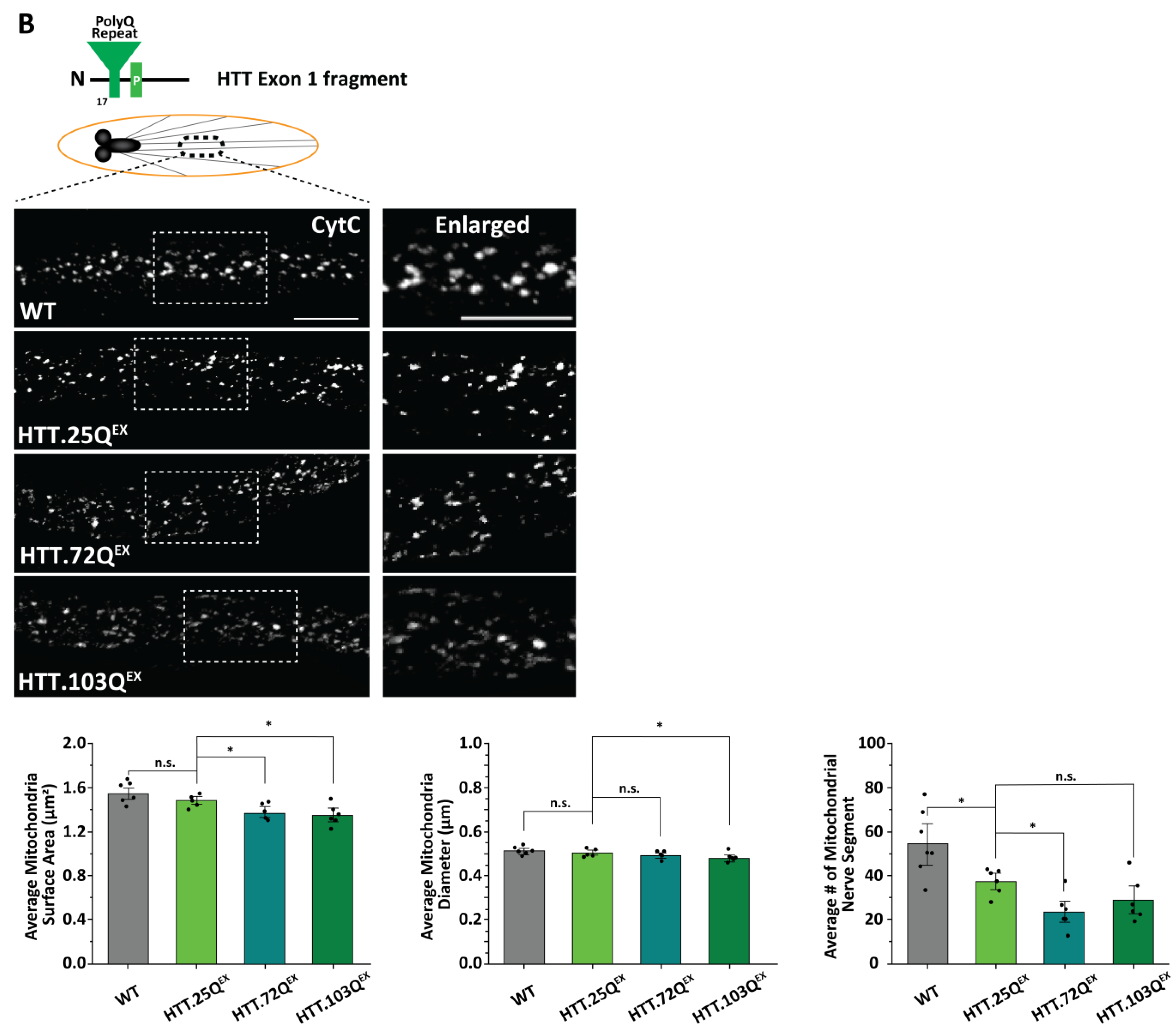
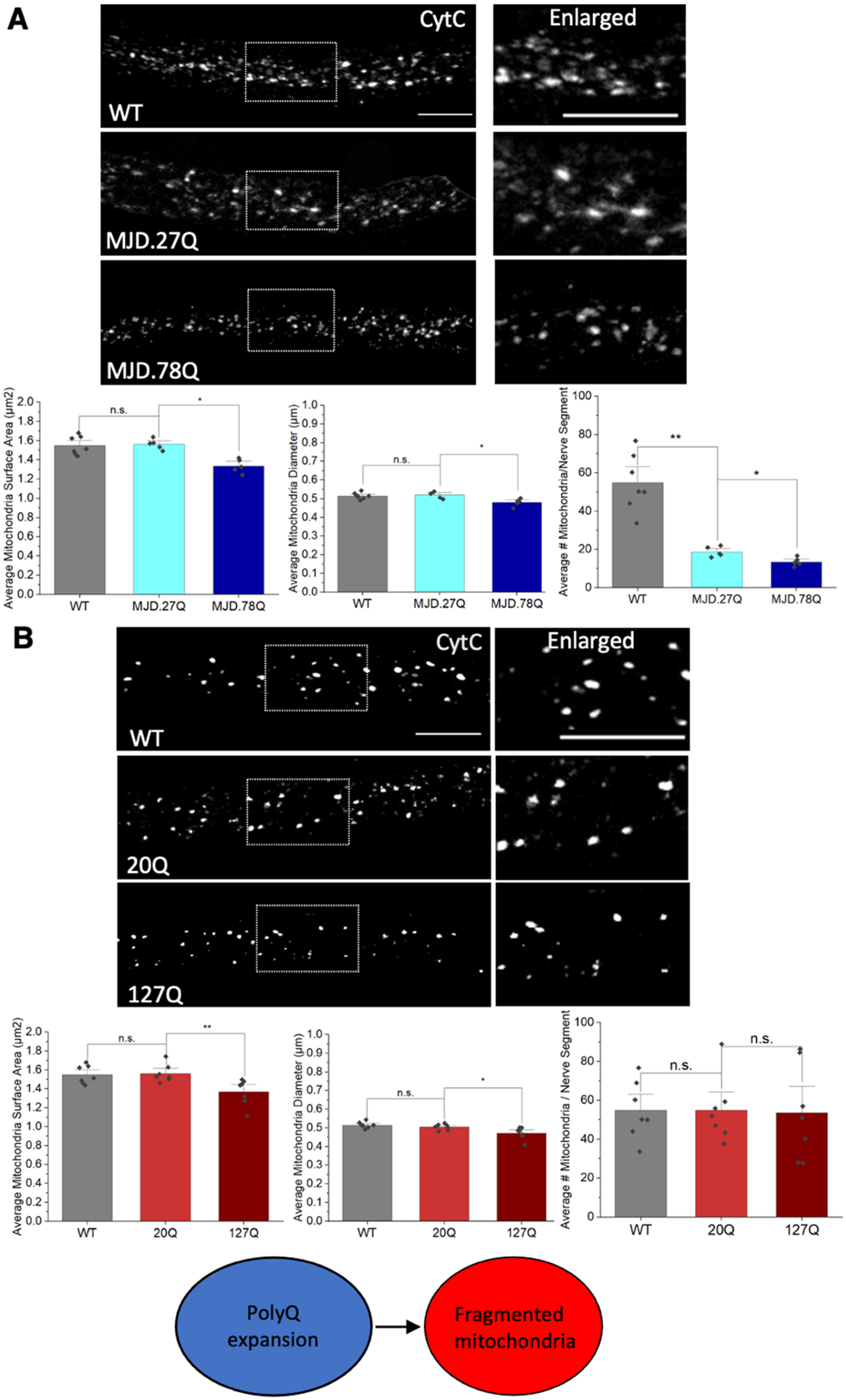
3.2. Expansion of PolyQ Repeats Alone Can Cause Mitochondria Fragmentation
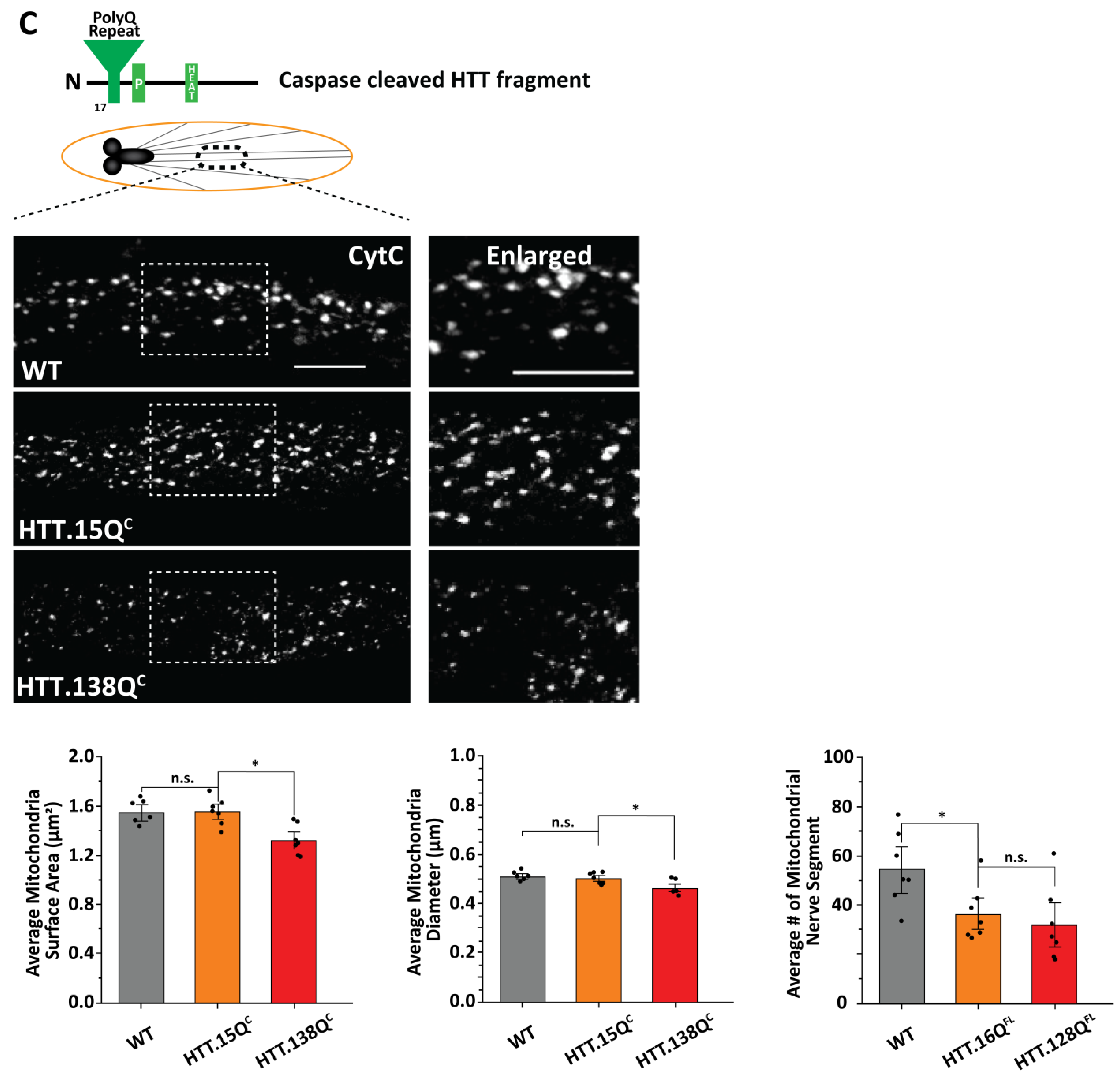
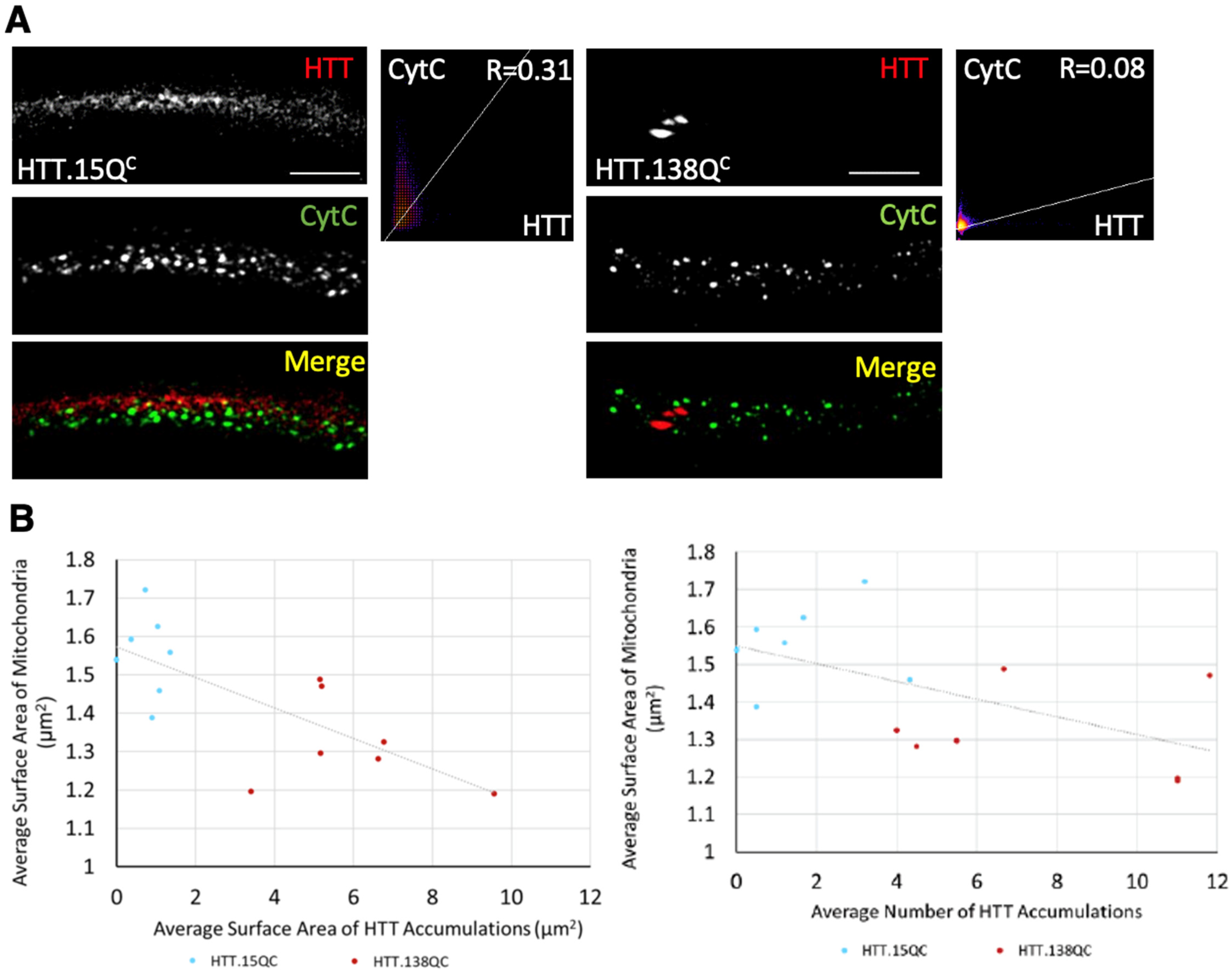
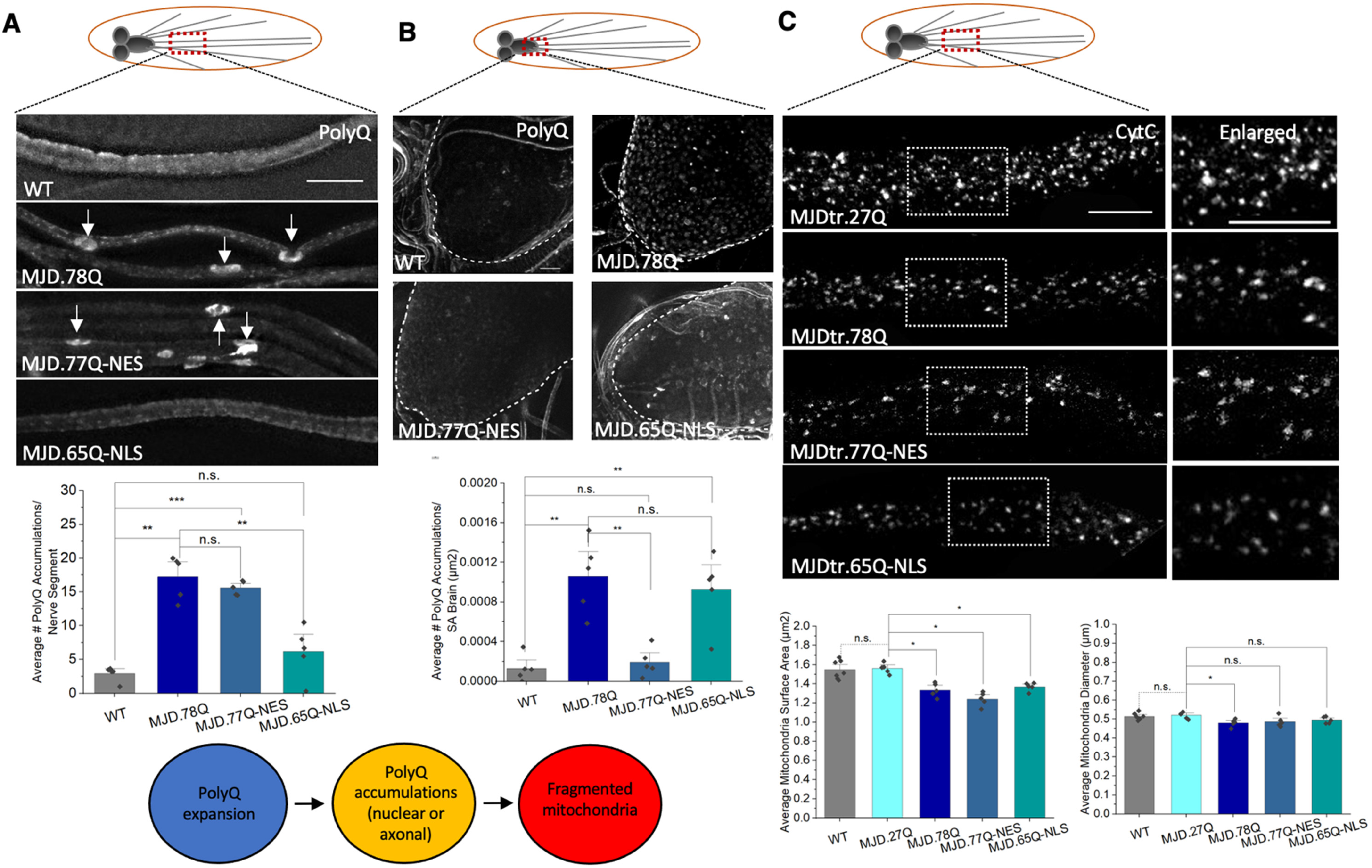
3.3. PolyQ-Mediated Mitochondria Fragmentation Is Not Dependent on the Cellular Location of polyQ Accumulations
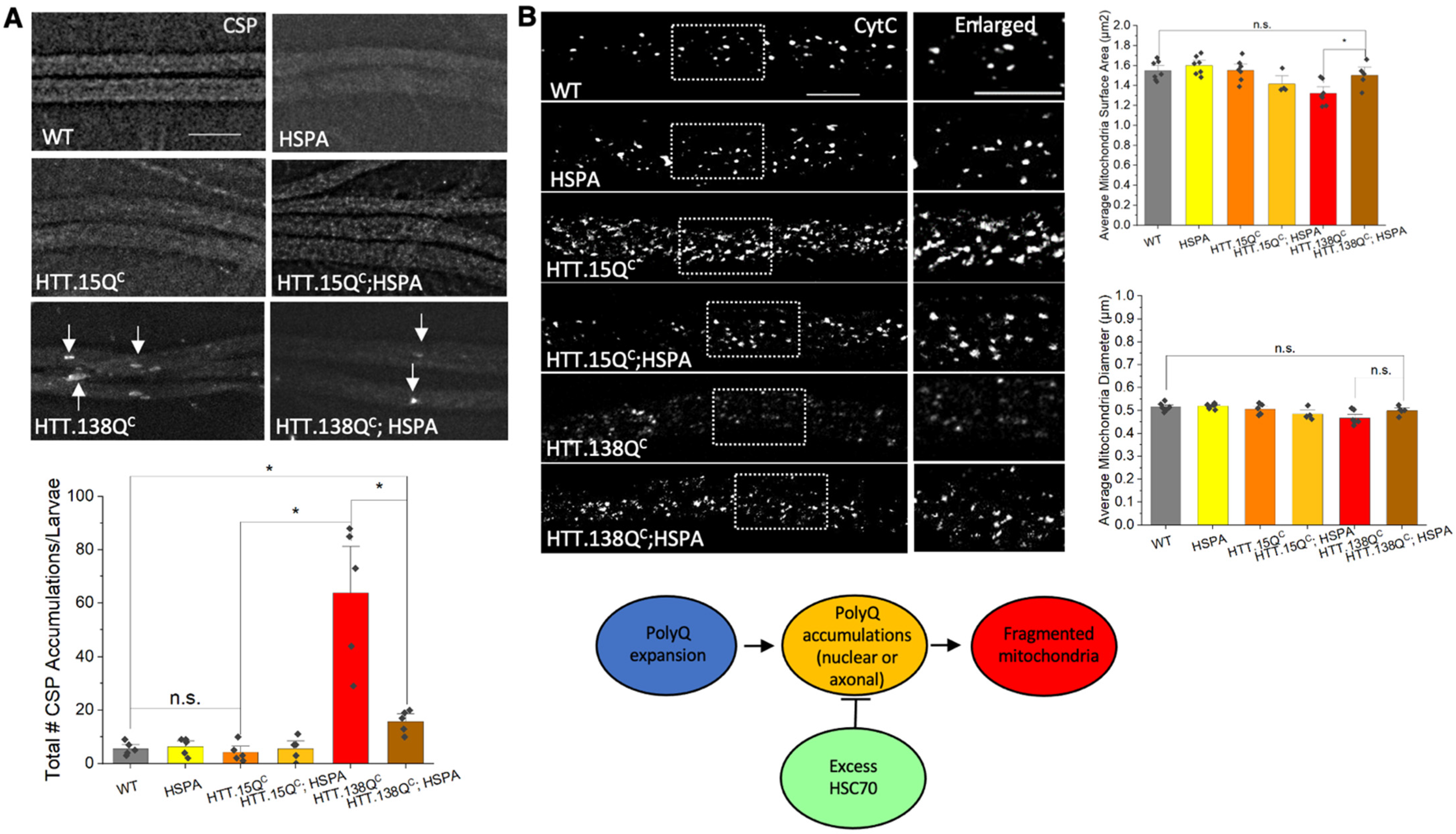
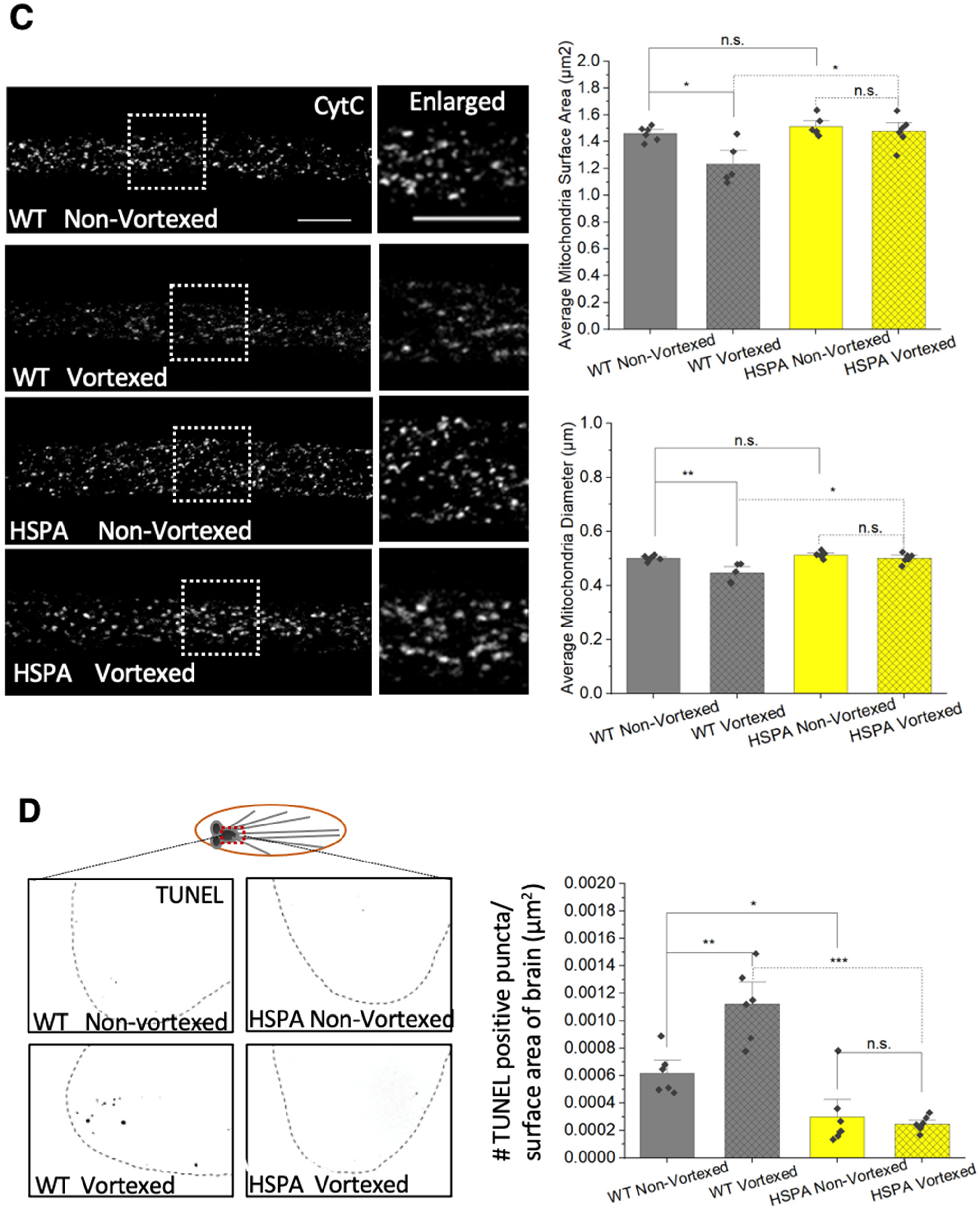
3.4. Expression of HSP70 Chaperone Protein Rescues PolyQ-Mediated Mitochondrial Fragmentation
3.5. PolyQ-Mediated Mitochondrial Fragmentation Is Not the Result of Cell Death
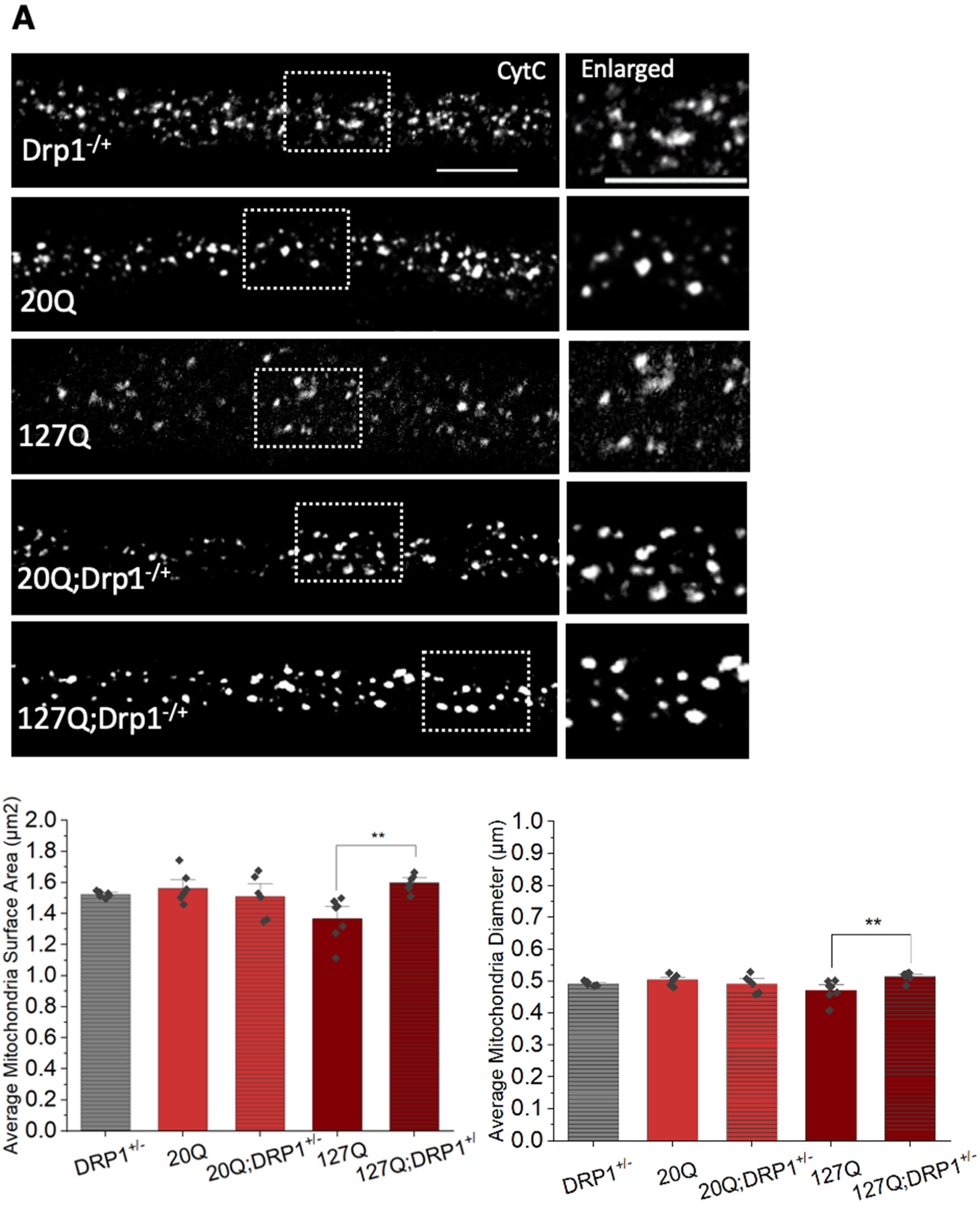
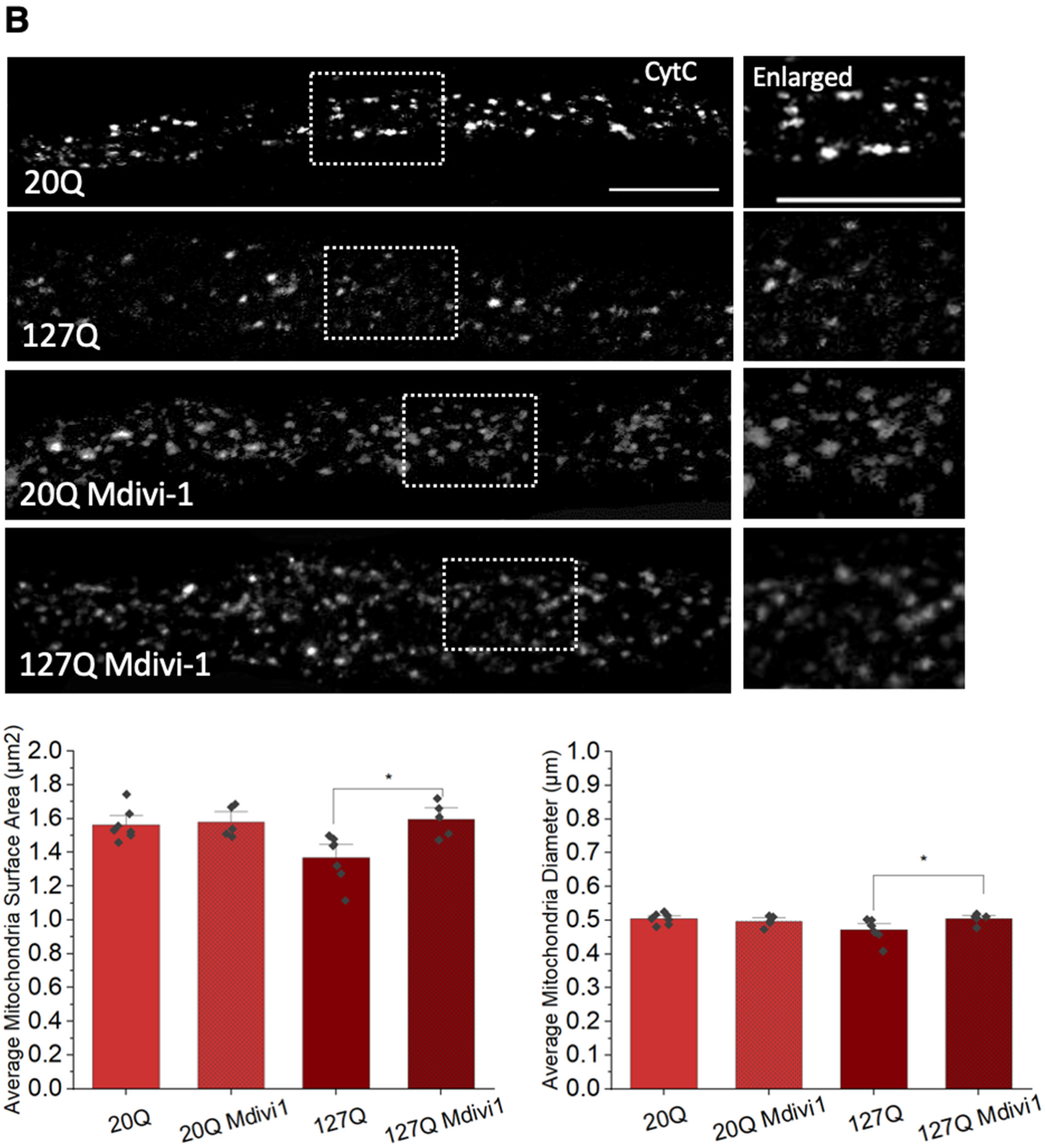
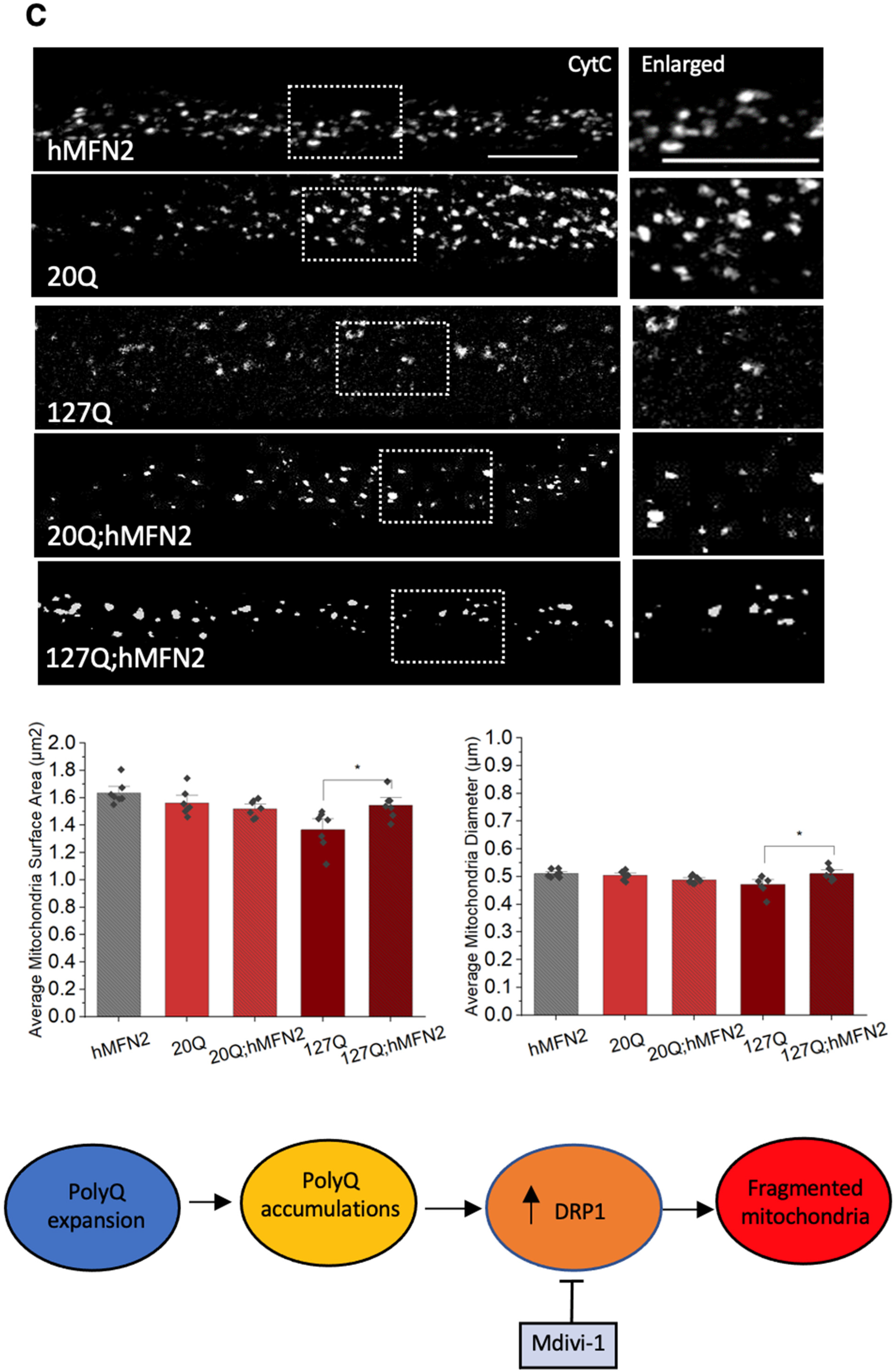
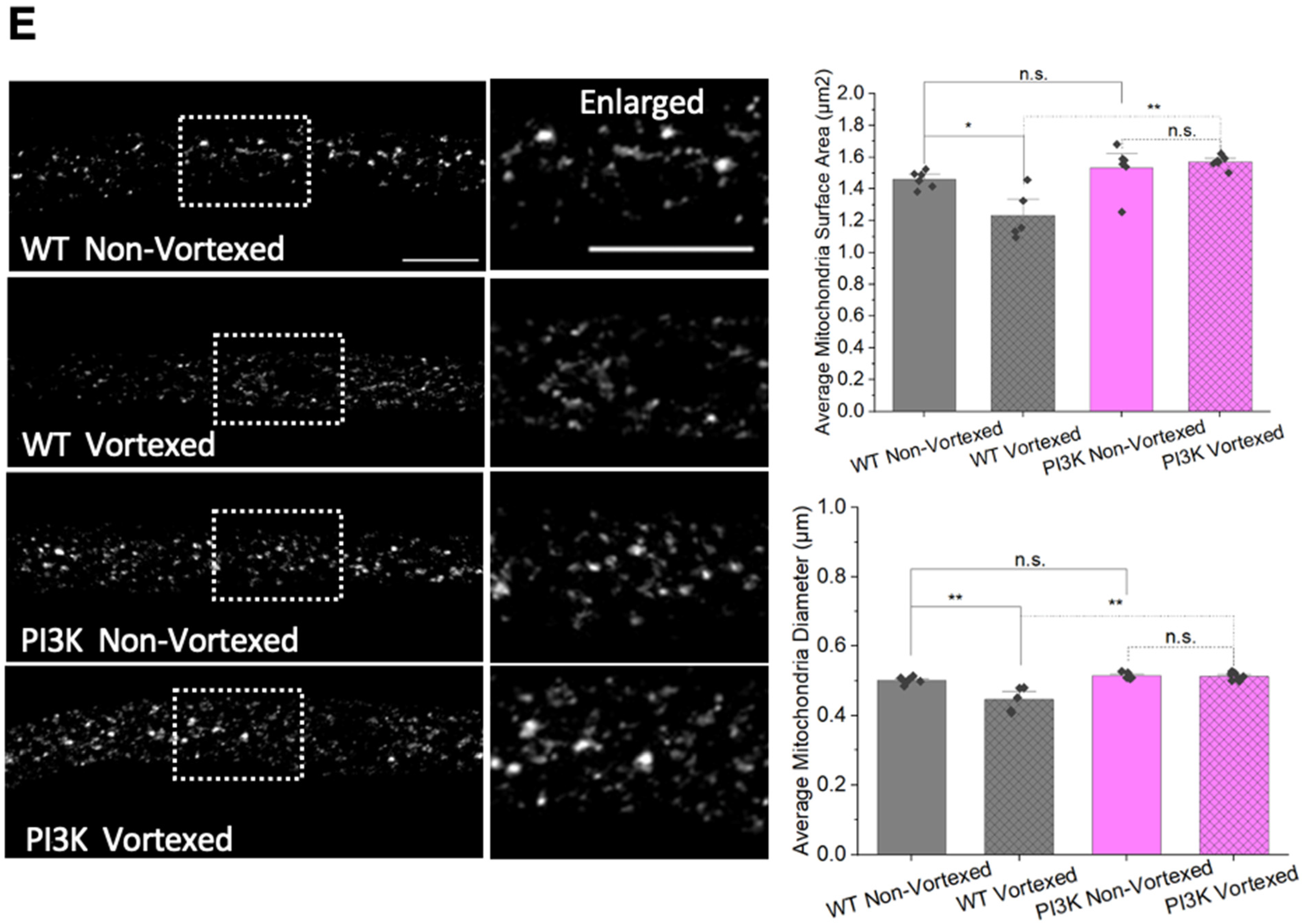
3.6. PolyQ-Mediated Mitochondria Fragmentation Is Caused by an Imbalance of Fission and Fusion Proteins
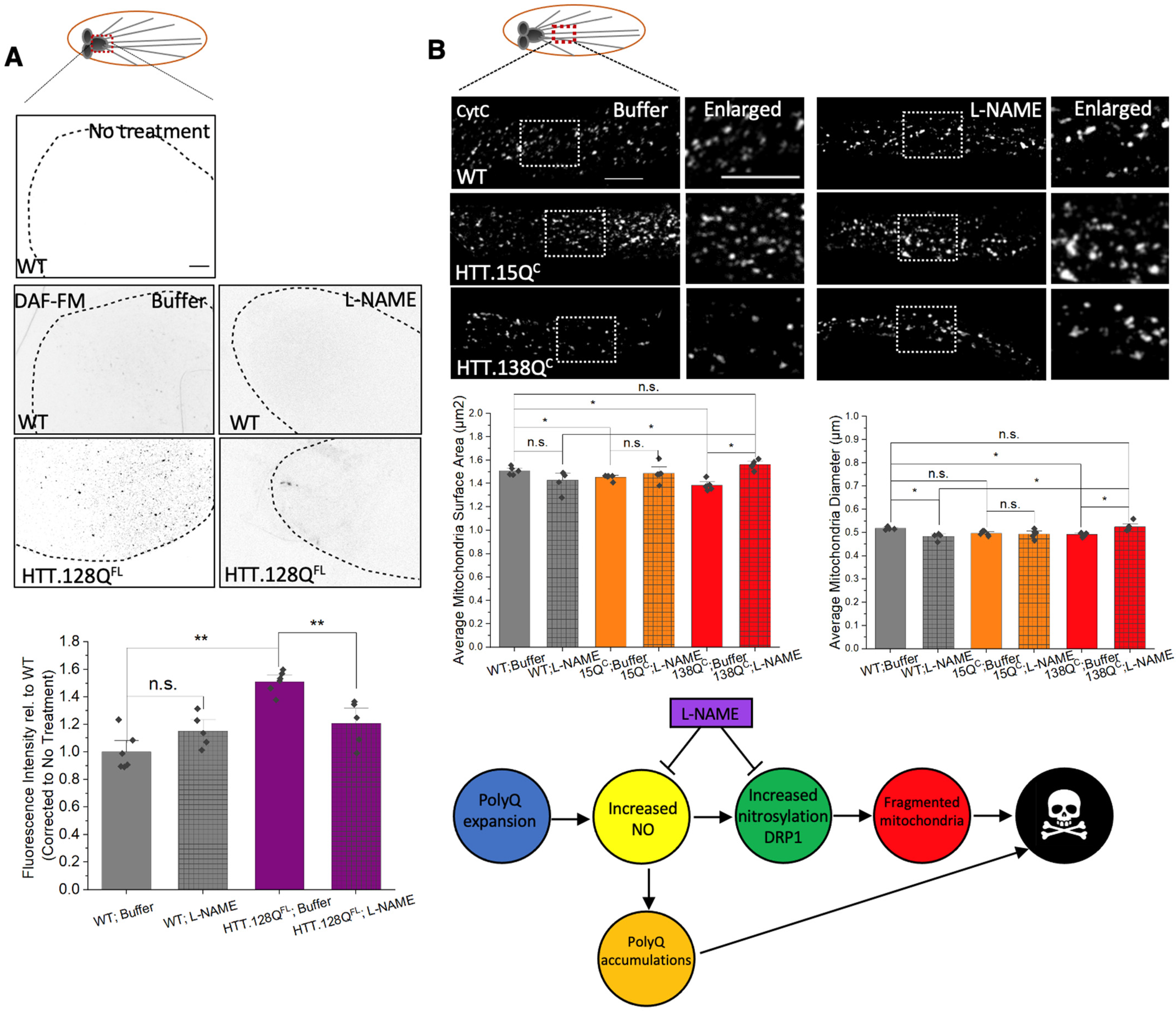
3.7. PolyQ-Mediated Mitochondria Fragmentation Is Likely Caused by Increased Levels of Nitric Oxide
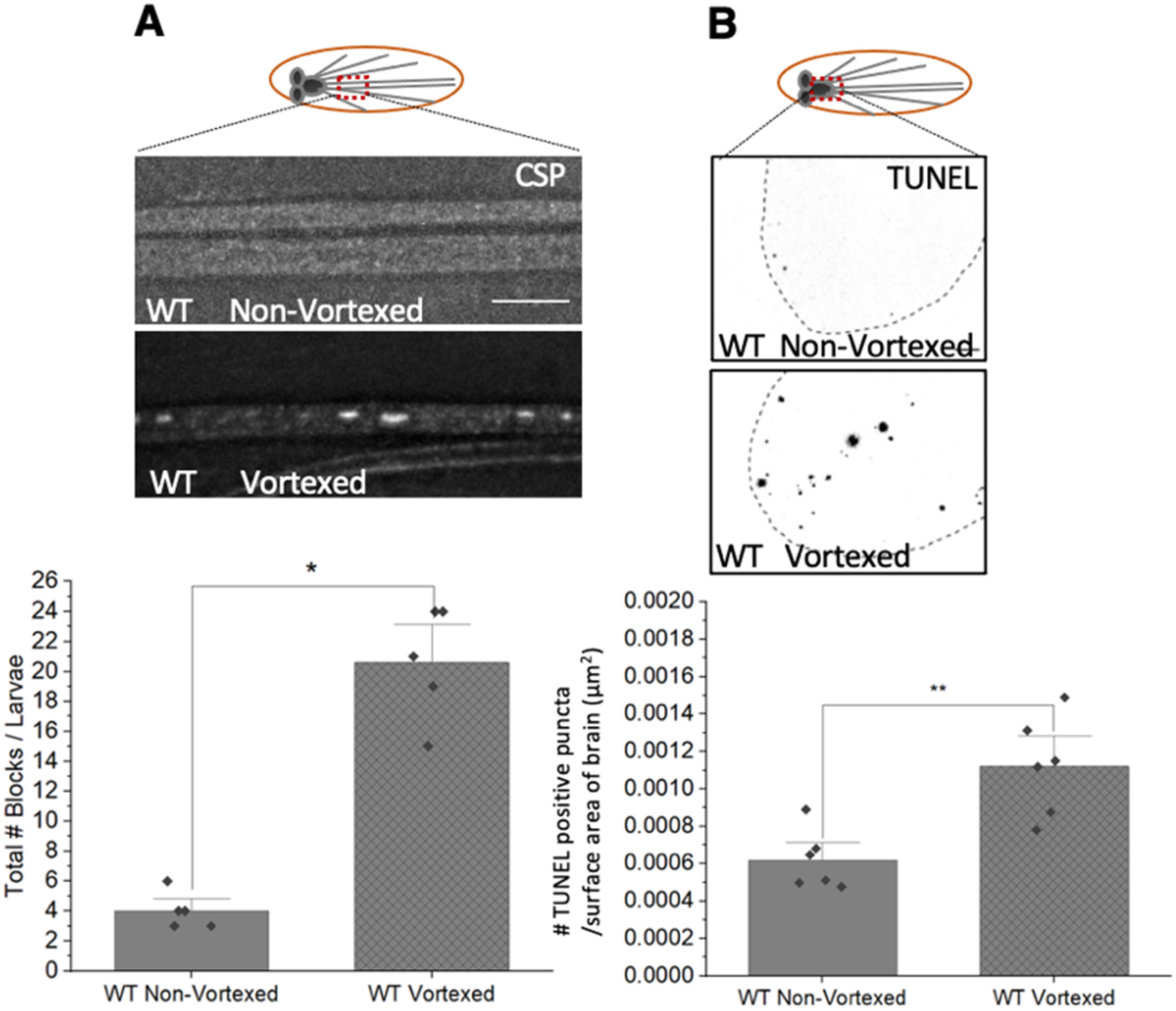
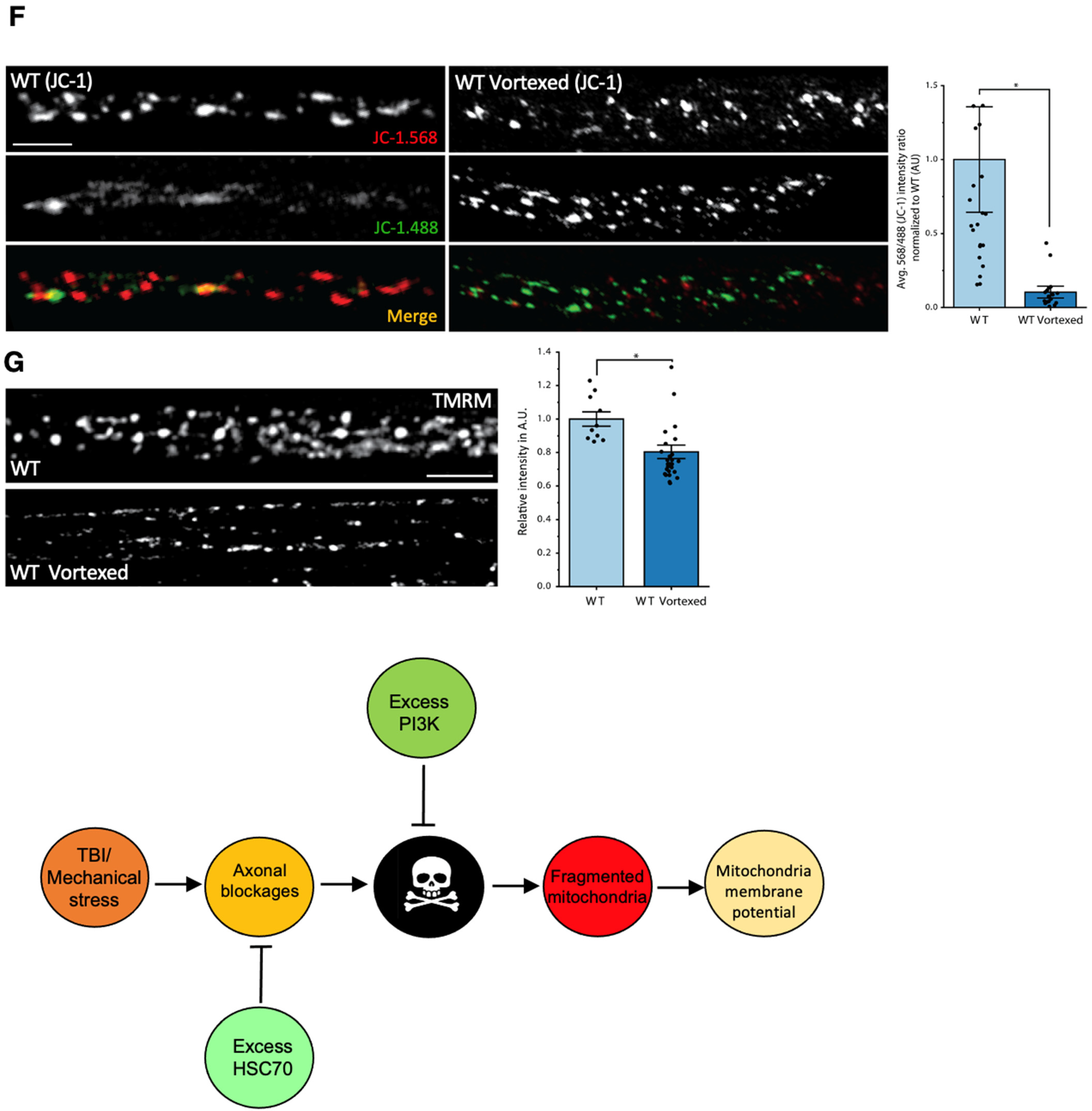
3.8. PolyQ-Mediated Mitochondrial Fragments Are Likely Not the Result of Fragmentation Caused by Mechanical Stress
4. Discussion
4.1. PolyQ Expansion Is Sufficient to Induce Mitochondrial Fragmentation Independent of HTT and Is Likely Not the Result of Cell Death
4.2. PolyQ-Mediated Mitochondria Fragmentation Is Distinct from TBI-Mediated Fragmentation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Key Resources Table
| Resource | Source | Identifier |
| Antibodies and Dyes | ||
| Mouse anti-Cytochrome C (7H8.2C12) | BD Pharmingen | Cat#556433 RRID: AB_396417 |
| Mouse anti-Polyglutamine | Fisher Scientific | Cat# MAB1574MI RRID: N/A |
| Rabbit anti-DRP1 | Laboratory of Leo Pallanck | Poole et al., 2010 [64] |
| Rabbit anti-Marf | Laboratory of Alex Whitworth | Zivani et al., 2010 [24] |
| Mouse anti-Tubulin (DM1A) | Abcam | Cat#ab7291 RRID: AB_2241126 |
| Anti-Mouse Alexa Fluor® 488 | Thermo Fisher | Cat# A1101 RRID: AB_2534069 |
| Anti-Mouse Alexa Fluor® 568 | Thermo Fisher | Cat# A1104 RRID: AB_2534072 |
| Anti-Mouse secondary antibody, HRP | Thermo Fisher | Cat# 32430 RRID: AB_1185566 |
| Anti-rabbit secondary antibody, HRP | Thermo Fisher | Cat# 32460 RRID: AB_1185567 |
| In situ Cell Death Detection Kit, Fluorescein | Roche | Cat#11684795910 Version #17 |
| DAF-FM Diacetate | Thermo Fisher | Cat# D23844 RRID: N/A |
| TMRM | Thermo Fisher | Cat# I34361 RRID: N/A |
| JC-1 | Cayman Chemical | Cat# 10009172 RRID: N/A |
| Chemicals, Peptides, and recombinant proteins | ||
| Protease inhibitor cocktail | Pierce | Cat# PIA32965 RRID: N/A |
| Phosphatase Inhibitor | Pierce | Cat# P188667 RRID: N/A |
| VectaShield Mounting Medium | Fisher | Cat# NC9265087 RRID: N/A |
| Mitochondrial Division Inhibitor, Mdivi-1 | Fisher Scientific | Cat# 47-585-610MG PubChem: 33825829 |
| L-NAME hydrochloride | Fisher Scientific | Cat# 06-651-00 PubChem: 135193 |
| Experimental Models: D. melanogaster organisms/strains | ||
| P{w[+m*]=Appl-GAL4.G1a}1, y [1] w[*] | Bloomington Drosophila Stock Center | BDSC: 32040 FlyBase: FBst0032040 |
| Appl-GAL4; T(2,3), CyO, TM6B, Tb1/Pin88k | Laboratory of Lawrence Goldstein | Gunawardena and Goldstein, 2001 [34] |
| UAS-HTT.15Q.mRFP | Laboratory of J Troy Littleton | Weiss et al., 2012 [13] |
| UAS-HTT.138Q.mRFP | Laboratory of J Troy Littleton | Weiss et al., 2012 [13] |
| w[1118]; P{w[+mC]=UAS-HTT.16Q.FL}F24/CyO | Bloomington Drosophila Stock Center | BDSC: 33810 FlyBase: FBst0033810 |
| w[1118]; P{w[+mC]=UAS-HTT.128Q.FL}f27b | Bloomington Drosophila Stock Center | BDSC: 33808 FlyBase: FBst0033808 |
| UAS-HTT.25Q-eGFP | Laboratory of Dr. Norbert Perrimon | Zhang et al., 2010 [15] |
| UAS-HTT.72Q-eGFP | Laboratory of Dr. Norbert Perrimon | Zhang et al., 2010 [15] |
| UAS-HTT.103Q-eGFP | Laboratory of Dr. Norbert Perrimon | Zhang et al., 2010 [15] |
| UAS-20Q | Laboratory of Seymour Benzer | Kazemi-Esfarjani and Benzer, 2002 [16] |
| UAS-127Q | Laboratory of Seymour Benzer | Kazemi-Esfarjani and Benzer, 2002 [16] |
| UAS-MJD.27Q | Laboratory of Nancy Bonini | Bonini, 1999 [17] |
| UAS-MJD.78Q | Laboratory of Nancy Bonini | Bonini, 1999 [17] |
| UAS-MJD.Q77-NES/Cyo | Laboratory of Nancy Bonini | Gunawardena et al., 2003 [18] |
| UAS-MJD.Q65-NLS/Cyo | Laboratory of Nancy Bonini | Gunawardena et al., 2003 [18] |
| w1118; P{UAS-Hsap\HSPA1L.W} 41.1 | Bloomington Drosophila Stock Center | BDSC: 7454 FlyBase: FBst0007454 |
| y1; P{SUPor-P} Drp1KG03815/CyO; ry506 | Bloomington Drosophila Stock Center | BDSC: 13510 FlyBase: FBst0013510 |
| w1118; P{UAS-hMFN2.D}29/TM3, Sb1 | Bloomington Drosophila Stock Center | BDSC: 59044 FlyBase: FBst0059044 |
| P{w[+mC]=UAS-Pi3K92E.CAAX}1, y[1] w[1118] | Bloomington Drosophila Stock Center | BDSC: 8294 FlyBase: FBst0008294 |
| w*; P{UAS-Drp1.D}3 | Bloomington Drosophila Stock Center | BDSC: 51647 FlyBase: FBst0051647 |
| UAS-Mitotimer | Bloomington Drosophila Stock Center | BDSC: 57323 FlyBase: FBst0057323 |
| UAS-Mito-roGFP2-Grx1 | Bloomington Drosophila Stock Center | BDSC: 67664 FlyBase: FBst0067664 |
| UAS-Mito-roGFP2-ORP1 | Bloomington Drosophila Stock Center | BDSC: 67667 FlyBase: FBst0067667 |
| Software/algorithms | ||
| ImageJ | Schneider et al., 2012 [65] https://imagej.net/. Accessed on 2 October 2023. | RRID: SCR_003070 |
| Metamorph/Metavue Imaging Software | Molecular Devices, Sunnyvale, CA, USA | RRID: SCR_002368 |
| Minitab 18 | https://www.minitab.com/en-us/. Accessed on 2 October 2023. | RRID: SCR_014483 |
| Microsoft Excel | https://www.microsoft.com/en-gb/. Accessed on 2 October 2023. | RRID: SCR_016137 |
| OriginLab/OriginPro | https://www.originlab.com/. Accessed on 2 October 2023. | RRID: SCR_014212 |
References
- Youle, R.J.; van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q. Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front. Cell. Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid. Redox Signal 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. Exaggerated mitophagy: A weapon of striatal destruction in the brain? Biochem. Soc. Trans. 2020, 48, 709–717. [Google Scholar] [CrossRef]
- Czeredys, M. Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington’s Disease. Front. Cell Dev. Biol. 2020, 8, 611735. [Google Scholar] [CrossRef]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium Homeostasis and Mitochondrial Dysfunction in Striatal Neurons of Huntington Disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef]
- Mason, R.P.; Casu, M.; Butler, N.; Breda, C.; Campesan, S.; Clapp, J.; Green, E.W.; Dhulkhed, D.; Kyriacou, C.P.; Giorgini, F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 2013, 45, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Imarisio, S.; Fleming, A.; Rose, C.; Krishna, G.; Heard, P.; Quick, M.; Korolchuk, V.I.; Renna, M.; Sarkar, S.; et al. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 2010, 19, 3413–3429. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.R.; Kimura, Y.; Lee, W.C.M.; Littleton, J.T. Huntingtin Aggregation Kinetics and Their Pathological Role in a Drosophila Huntington’s Disease Model. Genetics 2012, 190, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Cha, G.H.; Verstreken, P.; Ly, C.V.; Hughes, R.E.; Bellen, H.J.; Botas, J. Suppression of Neurodegeneration and Increased Neurotransmission Caused by Expanded Full-Length Huntingtin Accumulating in the Cytoplasm. Neuron 2008, 57, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Binari, R.; Zhou, R.; Perrimon, N. A Genomewide RNA Interference Screen for Modifiers of Aggregates Formation by Mutant Huntingtin in Drosophila. Genetics 2010, 184, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Esfarjani, P.; Benzer, S. Suppression of polyglutamine toxicity by a Drosophila homolog of myeloid leukemia factor 1. Hum. Mol. Genet. 2002, 11, 2657–2672. [Google Scholar] [CrossRef] [PubMed]
- Bonini, N.M. A genetic model for human polyglutamine-repeat disease in Drosophila melanogaster. Philos. Trans. R. Soc. B Biol. Sci. 1999, 354, 1057–1060. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of Axonal Transport by Loss of Huntingtin or Expression of Pathogenic PolyQ Proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Krzystek, T.J.; Banerjee, R.; Thurston, L.; Huang, J.; Swinter, K.; Rahman, S.N.; Falzone, T.L.; Gunawardena, S. Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis. 2021, 12, 796. [Google Scholar] [CrossRef]
- Xie, J.; Chen, S.; Bopassa, J.C.; Banerjee, S. Drosophila tubulin polymerization promoting protein mutants reveal pathological correlates relevant to human Parkinson’s disease. Sci. Rep. 2021, 11, 13614. [Google Scholar] [CrossRef]
- Wang, Z.H.; Clark, C.; Geisbrecht, E.R. Analysis of mitochondrial structure and function in the Drosophila larval musculature. Mitochondrion 2016, 26, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.X.; Lecoeur, H.; Zorn, S.E.; Dauguet, C.; Mignotte, B.; Gougeon, M.L. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 1995, 130, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1630–1639. [Google Scholar] [CrossRef]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Casale, A.M.; Liguori, F.; Ansaloni, F.; Cappucci, U.; Finaurini, S.; Spirito, G.; Persichetti, F.; Sanges, R.; Gustincich, S.; Piacentini, L. Transposable element activation promotes neurodegeneration in a Drosophila model of Huntington’s disease. iScience 2022, 25, 103702. [Google Scholar] [CrossRef]
- Hansen, T.; Thant, C.; White, J.A.; Banerjee, R.; Thuamsang, B.; Gunawardena, S. Excess active P13K rescues huntingtin-mediated neuronal cell deathbut has no effect on axonal transport defects. Apoptosis 2019, 24, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, B.A.; Lukacsovich, T.; Agrawal, N.; Burke, J.; Bornemann, D.J.; Purcell, J.M.; Worthge, S.A.; Caricasole, A.; Weiss, A.; Song, W.; et al. Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease. Hum. Mol. Genet. 2015, 24, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Krzystek, T.J.; White, J.A.; Rathnayake, R.; Thurston, L.; Hoffmar-Glennon, H.; Li, Y.; Gunawardena, S. HTT (huntingtin) and RAB7 co-migrate retrogradely on a signaling LAMP1-containing late endosome during axonal injury. Autophagy 2023, 19, 1199–1220. [Google Scholar] [CrossRef]
- Telenius, H.; Kremer, H.P.H.; Thellmann, J.; Andrew, S.E.; Almqvist, E.; Anvret, M.; Greenberg, C.; Greenberg, J.; Lucotte, G.; Squltierl, F.; et al. Molecular analysis of juvenile Huntington disease: The major influence on (CAG) n repeat length is the sex of the affected parent. Hum. Mol. Genet. 1993, 2, 1535–1540. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef]
- Tsou, W.L.; Burr, A.A.; Ouyang, M.; Blount, J.R.; Scaglione, K.M.; Todi, S.V. Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J. Biol. Chem. 2013, 288, 34460–34469. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.M.; Chan, H.Y.; Gray-Board, G.L.; Chai, Y.; Paulson, H.L.; Bonini, N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999, 23, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Sandoval, H.; Yao, C.K.; Chen, K.; Jaiswal, M.; Donti, T.; Lin, Y.Q.; Bayat, V.; Xiong, B.; Zhang, K.; David, G.; et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. eLife 2014, 3, e03558. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Satoh, A.; Chatterjee, S.; Toomre, D.K.; Chalouni, C.M.; Fulton, D.; Groszmann, R.J.; Shah, V.H.; Sessa, W.C. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA 2006, 103, 19777–19782. [Google Scholar] [CrossRef]
- Barekat, A.; Gonzalez, A.; Mauntz, R.E.; Kotzebue, R.W.; Molina, B.; El-Mecharrafie, N.; Conner, C.J.; Garza, S.; Melkani, G.C.; Joiner, W.J.; et al. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci. Rep. 2016, 6, 25252. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. Physiol. J. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Wellington, C.L.; Ellerby, L.M.; Gutekunst, C.A.; Rogers, D.; Warby, S.; Graham, R.K.; Loubser, O.; van Raamsdonk, J.; Yang, Y.Z.; Gafni, J.; et al. Caspase Cleavage of Mutant Huntingtin Precedes Neurodegeneration in Huntington’s Disease. J. Neurosci. 2002, 22, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the Caspase-6 Site Is Required for Neuronal Dysfunction and Degeneration Due to Mutant Huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Li, S.; Wang, C.E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Kegel, K.B.; Sapp, E.; Yoder, J.; Cuiffo, B.; Sobin, L.; Kim, Y.J.; Qin, Z.H.; Hayden, M.R.; Aronin, N.; Scott, D.L.; et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 2005, 280, 36464–36473. [Google Scholar] [CrossRef]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Liévens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749. [Google Scholar]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A mitochondria-associated oxidative stress perspective on Huntington’s disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Puranam, K.L.; Wu, G.; Strittmatter, W.J.; Burke, J.R. Polyglutamine expansion inhibits respiration by increasing reactive oxygen species in isolated mitochondria. Biochem. Biophys. Res. Commun. 2006, 341, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Wardman, J.H.; Hargreaves, I.P.; Neergheen, V.; Bie, A.S.; Tümer, Z.; Nielsen, J.E.; Nielsen, T.T. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion 2017, 34, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Gutiérrez-Ángel, S.; Schaefer, M.H.; Cox, J.; Meissner, F.; Hipp, M.S.; Hartl, F.U.; Klein, R.; Dudanova, I.; Mann, M. Spatiotemporal proteomic profiling of Huntington’s disease inclusions reveals widespread loss of protein function. Cell Rep. 2017, 21, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Ripolone, M.; Lucchini, V.; Ronchi, D.; Fagiolari, G.; Bordoni, A.; Fortunato, F.; Mondello, S.; Bonato, S.; Meregalli, M.; Torrente, Y.; et al. Purkinje cell COX deficiency and mtDNA depletion in an animal model of spinocerebellar ataxia type 1. J. Neurosci. Res. 2018, 96, 1576–1585. [Google Scholar] [CrossRef]
- Tichanek, F.; Salomova, M.; Jedlicka, J.; Kuncova, J.; Pitule, P.; Macanova, T.; Petrankova, Z.; Tuma, Z.; Cendelin, J. Hippocampal mitochondrial dysfunction and psychiatric-relevant behavioral deficits in spinocerebellar ataxia 1 mouse model. Sci. Rep. 2020, 10, 5418. [Google Scholar] [CrossRef]
- Ward, J.M.; Stoyas, C.A.; Switonski, P.M.; Ichou, F.; Fan, W.; Collins, B.; Wall, C.E.; Adanyeguh, I.; Niu, C.; Sopher, B.L.; et al. Metabolic and organelle morphology defects in mice and human patients define spinocerebellar ataxia type 7 as a mitochondrial disease. Cell Rep. 2019, 26, 1189–1202. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Jhang, Y.L.; Cheng, P.H.; Chang, Y.F.; Mao, S.H.; Yang, H.I.; Lin, C.W.; Chen, C.M.; Yang, S.H. The truncated C-terminal fragment of mutant ATXN3 disrupts mitochondria dynamics in spinocerebellar ataxia type 3 models. Front. Mol. Neurosci. 2017, 10, 196. [Google Scholar] [CrossRef]
- Deckel, A.W.; Tang, V.; Nuttal, D.; Gary, K.; Elder, R. Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington’s disease transgenic mice. Brain Res. 2002, 939, 76–86. [Google Scholar] [CrossRef]
- Cherian, L.; Hlatky, R.; Roberston, C. Nitric Oxide in Traumatic Brain Injury. Brain Pathol. 2004, 14, 195–201. [Google Scholar] [CrossRef]
- Villalba, N.; Sonkusare, S.K.; Longden, T.A.; Tran, T.L.; Sackheim, A.M.; Nelson, M.T.; Wellman, G.C.; Freeman, K. Traumatic brain injury disrupts cerebrovascular tone through endothelial inducible nitric oxide synthase expression and nitric oxide gain of function. J. Am. Heart Assoc. 2014, 3, e001474. [Google Scholar] [CrossRef]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Vidwans, A.S.; Kim, S.; Coffin, D.O.; Wink, D.A.; Hewett, S.J. Analysis of the neuroprotective effects of various nitric oxide donor compounds in murine mixed cortical cell culture. J. Neurochem. 1999, 72, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Zambrano, A.; Barrientos, A. Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. 2010, 24, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]

















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swinter, K.; Salah, D.; Rathnayake, R.; Gunawardena, S. PolyQ-Expansion Causes Mitochondria Fragmentation Independent of Huntingtin and Is Distinct from Traumatic Brain Injury (TBI)/Mechanical Stress-Mediated Fragmentation Which Results from Cell Death. Cells 2023, 12, 2406. https://doi.org/10.3390/cells12192406
Swinter K, Salah D, Rathnayake R, Gunawardena S. PolyQ-Expansion Causes Mitochondria Fragmentation Independent of Huntingtin and Is Distinct from Traumatic Brain Injury (TBI)/Mechanical Stress-Mediated Fragmentation Which Results from Cell Death. Cells. 2023; 12(19):2406. https://doi.org/10.3390/cells12192406
Chicago/Turabian StyleSwinter, Kelsey, Dania Salah, Rasika Rathnayake, and Shermali Gunawardena. 2023. "PolyQ-Expansion Causes Mitochondria Fragmentation Independent of Huntingtin and Is Distinct from Traumatic Brain Injury (TBI)/Mechanical Stress-Mediated Fragmentation Which Results from Cell Death" Cells 12, no. 19: 2406. https://doi.org/10.3390/cells12192406