
In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy




1
Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA
2
Human Molecular Genetics and Physiology Program, Stanley Manne Children’s Research Institute at Ann & Robert H. Lurie Children’s Hospital, Chicago, IL 60611, USA
3
Department of Pediatrics, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
*
Author to whom correspondence should be addressed.
Brain Sci. 2021, 11(2), 194; https://doi.org/10.3390/brainsci11020194
Submission received: 2 December 2020
/
Revised: 29 January 2021
/
Accepted: 1 February 2021
/
Published: 5 February 2021
(This article belongs to the Special Issue A Decade of Brain Sciences)
Abstract
:Until the recent development of disease-modifying therapeutics, spinal muscular atrophy (SMA) was considered a devastating neuromuscular disease with a poor prognosis for most affected individuals. Symptoms generally present during early childhood and manifest as muscle weakness and progressive paralysis, severely compromising the affected individual’s quality of life, independence, and lifespan. SMA is most commonly caused by the inheritance of homozygously deleted SMN1 alleles with retention of one or more copies of a paralog gene, SMN2, which inversely correlates with disease severity. The recent advent and use of genetically targeted therapies have transformed SMA into a prototype for monogenic disease treatment in the era of genetic medicine. Many SMA-affected individuals receiving these therapies achieve traditionally unobtainable motor milestones and survival rates as medicines drastically alter the natural progression of this disease. This review discusses historical SMA progression and underlying disease mechanisms, highlights advances made in therapeutic research, clinical trials, and FDA-approved medicines, and discusses possible second-generation and complementary medicines as well as optimal temporal intervention windows in order to optimize motor function and improve quality of life for all SMA-affected individuals.
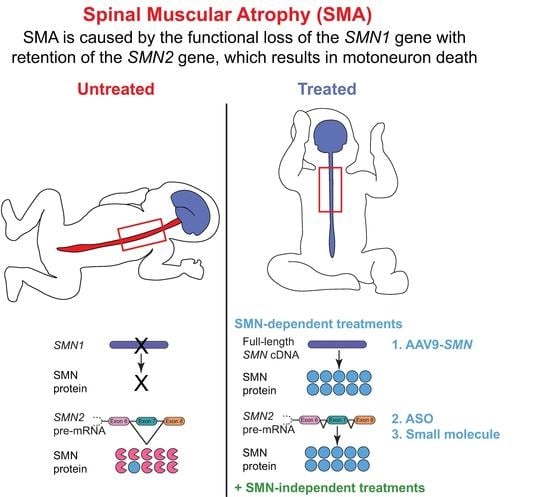
1. Genetics
Over the last 129 years, physicians and researchers have made substantial progress in recognizing, understanding, and treating the autosomal recessive genetic disorder spinal muscular atrophy (SMA). Although universally distinguishable by the pathological loss of lower α-motoneurons (most often during infancy or adolescence), SMA-affected individuals can present with a clinically heterogenous spectrum of symptoms. The variable presentation of symptoms undoubtedly obfuscated the recognition of the singular and unifying etiology of this disorder for the first 100 years after initial documentation. Generally, motor symptoms appear as symmetrical, predominantly proximal and axial muscle weakness and range from fatal somatic and respiratory paralysis to minor gait abnormalities. In contrast to the diverse presentation of this disorder, SMA is simply a complex manifestation of a relatively straightforward problem.
SMA is a monogenic, autosomal recessive disorder caused by the homozygous functional loss or deletion of a 5q13 gene critical for viability of motoneurons (aptly named survival motor neuron, or specifically in higher primates, SMN1) [1]. The absence of SMN protein results in embryonic lethality [2]; thus, SMN is an essential protein for development. A single copy of the SMN gene is phylogenetically conserved across most eukaryotes, but humans are uniquely fortunate to possess a second SMN gene. The range in clinical symptoms is generally attributed to variable expression of this genetic paralog, survival motor neuron 2 (SMN2). The paramount difference in the nucleotide sequence of SMN1 versus SMN2 is a translationally silent cytosine (C) to thymine (T) nucleotide transition at the sixth position in SMN2 exon 7 [3,4]. This nucleotide transition modifies SMN2 pre-mRNA splicing and promotes exon 7 exclusion from the mature transcript (Figure 1). SMN2 exon 7 skipping occurs because the C to T transition impedes an exonic splicing enhancer (ESE) site to which SF2/ASF binds [5] and creates a novel exonic splicing silencer (ESS) site to which the splicing factors hnRNP A1/A2 bind [6]. The alternatively spliced SMNΔ7 protein is unstable, inefficiently self-oligomerizes and is subsequently degraded [7]. While SMN1 selectively generates full-length SMN protein, only ~10 percent of protein produced by SMN2 is full-length SMN, and the remaining 90 percent is the less-functional SMNΔ7 splice variant that is rapidly eliminated. Due to the low production of functional SMN protein, SMN2 can only partially compensate for loss of SMN1, and thus SMA arises from a deficiency but not complete depletion of SMN protein.
Curiously, SMN2 copy number varies in the human population and ranges from 0 to 8 copies. This variability is likely due to the characteristic instability of the chromosome 5q13 region [8]. Each extra SMN2 copy at the SMN locus serves to bolster the amount of functional, full-length SMN protein generated within a cell. This natural variability in SMN2 expression in the absence of SMN1 permits the clinical SMA severity to exist along a spectrum predominantly determined by SMN2 copy number [9,10,11,12].
2. Clinical Manifestations
Clinical features that define SMA include progressive muscle weakness, hypotonia, and atrophy of skeletal muscle resulting from degeneration and loss of spinal motoneurons [13,14]. Given the wide range of clinical phenotypes, SMA has been traditionally classified into five categories (Type 0 to Type IV) based on age at symptom onset and highest motor milestone achieved (Table 1) [15,16,17,18]. Generally, the clinical subtype inversely correlates with SMN2 copy number and may provide physicians with some clinical insight and prognostic information.
The natural history of each clinical subtype (reviewed in [19]) is described as follows: in all subtypes, proximal muscle groups are more affected than distal muscle groups, with legs more affected than arms. The most severe form of SMA (Type 0) may present with prenatal onset (reduced movement in utero) and newborns may be born unable to swallow and breathe independently. These infants exhibit severe muscle weakness and lack of tone, joint contractures, areflexia, bilateral facial paralysis and early respiratory failure [20,21]. Outside of bulbar and spinal motoneuron dysfunction, thalamus, heart, vasculature, and sensory system may also develop problems due to exceedingly low systemic SMN levels [22,23,24,25,26]. Type 0 SMA patients carry a single copy of SMN2 and have life expectancies of less than six months.
Approximately half of all SMA cases are classified as Type I (Werdnig–Hoffman disease). Infants with Type I SMA exhibit symptom onset prior to six months of age and are unable sit unassisted due to proximal muscle weakness and hypotonia [27]. This weakness manifests as a frog-leg posture, poor head control, paradoxical breathing, bulbar involvement (tongue fasciculation and dysphagia), reduced or absent reflexes, and respiratory failure before the age of two. Without respiratory, nutritional, or genetically targeted interventions, life expectancy is less than two years of age [28]. Type I SMA patients typically have two copies of SMN2. Although Type I SMA has the highest incidence of the five clinical subtypes, milder forms of the disorder are historically more prevalent in the population due to the increased lifespan of intermediate and mild SMA patients (Type II–IV). However, the advent and use of genetically targeted therapies for SMA will dramatically shift this distribution.
Patients with Type II SMA (Dubowitz disease) manifest motor symptoms between six and 18 months of age. These patients may sit without assistance but do not independently stand or achieve ambulation [29]. Type II SMA patients display progressive proximal muscle weakness, lack muscle tone, and present with diminished reflexes. Respiratory dysfunction is common, and the development of scoliosis plus weakened intercostal muscles can affect pulmonary function. Type II SMA patients develop hand tremors (polyminimyoclonus), contractures, and occasionally ankylosis of the mandible [13]. These patients typically have three copies of SMN2 and reduced life expectancy.
Patients with Type III SMA (Kugelberg–Welander disease) experience initial symptom onset during early childhood but substantial loss of ambulation generally occurs during puberty [30]. These individuals present with progressive proximal muscle weakness that is greater in the legs than the arms, and may retain the ability to walk but with considerable difficulty due to gait abnormalities and muscle atrophy/weakness [19]. However, some of these patients irreversibly lose walking ability. Type III SMA patients may develop polyminimyoclonus, but respiratory dysfunction and severe scoliosis are not components of the clinical description. Patients typically have 3–4 copies of SMN2 and a normal or near-normal life expectancy.
Lastly, patients with Type IV SMA (<5% of cases) develop symptoms in adulthood and experience the mildest disease course (generally restricted to gait abnormalities), which is attributed to them usually having ≥ 4 SMN2 copies [18,31]. While clinical subcategorization of SMA is useful in guiding treatment strategies and management of medical and developmental expectations, it is crucial to recognize that the clinical spectrum of SMA severity exists on a continuum and that division into subtypes may not fully capture the experience and expectations of patients, their families, and their health care providers. In addition, the recent advancements in disease-modifying SMA therapies have substantially altered the expectations of categorical patients, resulting in patients transcending subtypes. As SMN-dependent and -independent therapies continue to improve and become widely accessible to SMA patients, traditional classifications will need to be improved to capture previously unidentifiable symptoms and outcome expectations revealed by prolonged lifespans and improved motor function.
3. Impact of Motor Impairment on Quality of Life
SMA-affected individuals experience a slow and progressive decline in motor function and report limitations arising from difficulties in mobility, daily activity, and pervasive fatigue associated with deteriorating physical health [32,33]. Though the initial severity of SMA symptoms (and thus the maximal motor functions achieved) is variable, depreciation of function results in considerable physical disability for all SMA-affected individuals. Inadequate motor function may require daily tasks to be performed by a caregiver in order to help maintain or enhance an individual’s autonomy. These tasks include activities such as meal preparation and hygiene maintenance and are usually informally provided by relatives. One Spanish study evaluating the economic burden incurred by SMA-affected individuals and their caregivers found that the invisible “cost” (compared to medical cost) of informal caregiving accounts for more than two-thirds of the total annual cost of healthcare associated with SMA [34]. This cost is one of the reasons why parents of individuals with SMA declare that a critical gap in patient needs is the lack of support for activities associated with daily living [35].
In addition to motor impairment, patients also report emotional difficulties, which thus far lack effective interventions. These difficulties are due in part to the immense psychosocial burdens experienced by those living with SMA. In addition to the need to make difficult treatment choices, stress, limitations on social activities, and a lack of independence, the pervasive fear of losing functional ability significantly contributes to substantial negative mental health experienced by patients and their families [36]. When surveyed on which tasks would meaningfully improve quality of life, SMA patients reported that improved ability to do daily tasks such as eating, bathing, grooming, using the restroom, independent transfers to and from wheelchairs, being able to spend time independently, and typing on keyboards would be immensely beneficial [37]. The ability to independently perform these often-underappreciated motor tasks should be a goal for the treatment of SMA, and small changes may be sufficient to significantly improve quality of life. Patient, caregiver, and clinician perspectives indicate that even minor improvements in motor function would constitute a meaningful change in disorder outcome [38].
Until the recent development of genetic therapies, management of SMA was traditionally limited to long-term and multi-disciplinary medical, nutritional, and supportive care [39,40,41,42] to alter the natural disorder progression. The advancement of genetic therapies is reshaping the therapeutic environment by mitigating the need for invasive ventilation and extensive medical care, ameliorating progressive motor degeneration, and extending lifespan. However, key outcomes of clinical trials and therapeutic use indicate that impairments in motor function persist in some patient populations [43,44,45,46,47,48,49]. Additionally, not all patient populations are currently able to access or receive genetic therapies. Advances in supportive and therapeutic care have changed the diagnosis of SMA from devastating to hopeful, but complementary treatment approaches are required to further improve the quality of life experienced by SMA-affected individuals and their caregivers.
4. The SMN Protein
The SMN protein is ubiquitously expressed in eukaryotic cells, with the highest levels of expression in brain, liver, kidney, and spinal cord cells and moderate levels of expression in cardiac and skeletal muscle cells [50,51]. The full-length human SMN protein is comprised of 294 amino acids, has a molecular weight of 38 kDa, and contains four protein domains (reviewed in [52]): (1) an N-terminal lysine-rich domain (encoded by exons 2A and 2B) responsible for GEMIN2 and nucleic acid binding; (2) a central Tudor domain (encoded by exon 3) that mediates numerous interactions with arginine-glycine (RG)-rich proteins such as one family of core small nuclear ribonucleoproteins (snRNPs) called “Sm” proteins [53] (named by their reactivity with autoantibodies of the Sm serotype from patients with systemic lupus erythematosus [54]); (3) a C-terminal proline-rich domain (encoded by exons 4–6) that is responsible for binding profilin proteins; and (4) a tyrosine-glycine (YG) box (encoded by exon 6) that, along with the last sixteen residues encoded by exon 7, mediates SMN self-oligomerization [7]. Most of these protein domains are conserved amongst vertebrates. Interestingly, it was recently shown that removal of SMN exon 2B can result in a functional SMN protein that restores snRNP function and rescues cell lethality [55]. This implies that GEMIN2 binding is further refined to SMN exon 2A [55].
SMN exhibits both cytoplasmic and nuclear subcellular localization where the protein serves several proposed functions (Figure 2). SMN is a crucial component of snRNP biogenesis [56,57,58]. snRNPs are RNA–protein complexes that are critical constituents of spliceosomes, which recognize and remove introns from pre-mRNA. Cytoplasmic SMN acts in conjunction with GEMIN2-8 and UNR-interacting protein (UNRIP) as the SMN complex to load Sm proteins into a heptameric ring structure on small nuclear RNA (snRNA) [57,59,60,61,62,63,64,65,66,67,68,69]. After further snRNP processing, the SMN complex (still associated with the snRNP molecule) binds snurportin and importin and is transported into the nucleus. The SMN complex and associated snRNP molecules localize to Cajal bodies where snRNP maturation is completed. Other studies suggest that SMN may have numerous additional functions in RNA metabolism, protein homeostasis, and cytoskeletal dynamics (reviewed in [52,70,71]).
While the canonical and most defined role of SMN is snRNP assembly in the soma, several studies have proposed additional, non-canonical roles outside of the soma. In axons, SMN modulates axiogenesis and axonal pathfinding [72], and is involved in axonal transport of mRNA and synaptic vesicles [75,76,77,78]. At the synapse, SMN is involved in local translation of cytoskeletal proteins within presynaptic compartments [78,79]. At the growth cone and synaptic terminal of motoneurons, SMN regulates endocytosis and cytoskeleton activity through its interaction with profilins, a family of proteins regulating actin dynamics [78,80,81,82] and through this interaction has been proposed to regulate the change in ratio of G-actin to F-actin [83], a critical feature for neurite outgrowth [84]. After neuromuscular synapses have matured, SMN contributes to compensatory axonal sprouting following motor nerve injury [85]. It is prudent to note that it remains unclear how these proposed, non-canonical roles of SMN potentially contribute to SMA pathogenesis and symptomology, and more investigation is necessary to definitively understand the extent to which, if any, these potential non-splicing functions of SMN influence the SMA phenotype.
Although additional function(s) of SMN beyond its role in spliceosomal assembly are still debated, the fact that spinal motoneurons are preferentially susceptible to degeneration and death in response to SMN deficiency is quite clear. This has been firmly established through a series of genetic studies in which Smn levels are either specifically reduced or increased within motoneurons [86,87,88,89,90]. However, molecular mechanisms explaining the preferential vulnerability of spinal motoneurons to SMN deficiency are unknown. Three major hypotheses have been posited [71,72,91]. First, SMN deficiency may disrupt splicing of transcripts specifically essential for the function and survival of spinal motoneurons. This notion is supported by studies demonstrating that SMN deficiency differentially alters the snRNA composition of snRNPs across tissue types, which leads to tissue-specific patterns of alternative splicing [92,93]. Second, of all cell types, spinal motoneurons may be most sensitive to systemic splicing defects induced by SMN deficiency. It is possible that the requirement for SMN is uniquely high in spinal motoneurons, and thus even small reductions in SMN result in a deficiency leading to preferential vulnerability to degeneration. Third, SMN may have special spinal motoneuron-specific function(s) that render these cells particularly susceptible to neurodegeneration as a consequence of SMN deficiency.
5. Temporal Requirements of SMN
In humans and mammals, SMN expression levels are highest during embryonic and early postnatal development, sharply followed by a decrease to a basal level that is maintained throughout life (Figure 3) [94,95]. However, the complex mechanisms regulating this dynamic expression are not well understood. Peak SMN protein levels in the spinal cord are highest during the developmental window of axon sprouting from the spinal cord during early embryogenesis [96,97] and coincide with the onset of myelination [94]. In mice, maximal SMN expression occurs during embryonic day (E)10–13 to permit the growth and pathfinding of motoneuron growth cones to contact target muscles [97]. SMN undergoes an initial decline in expression between E14 and E19, after the developmental period of motoneuron innervation of muscle fiber endplates [98]. A secondary decline occurs between postnatal day (P)5 and P15 [99], which coincides with the time frame of neuromuscular junction (NMJ) maturation and stabilization.
Transgenic and conditional transgenic mouse models of SMA have greatly elucidated temporal and spatial requirements for SMN. For a detailed review of SMA animal models and their uses, we refer readers to Burghes et al. (2017) [100]. Briefly, increasing neuronal SMN levels (4-fold by E15) using a prion (PrP)-SMN cDNA transgene corrects the SMA phenotype seen in severe SMA mice [101], suggesting that elevated neuronal SMN expression is required during the perinatal developmental period. Indeed, restoring Smn expression during the early symptomatic period (P4-P8, but not P10) rescues neuromuscular pathology and motor function in mice co-expressing a ubiquitous Cre transgene and Smn-inducible alleles (SmnRes/Res) on the severe SMNΔ7 SMA background [95]. Le et al. (2011) were the first group to demonstrate the phenomenon that earlier SMN induction is most effective [102]. A tetracycline inducible expression system on the SMNΔ7 SMA mouse background was used to rescue SMN levels in SMA mice during both embryonic (E13) and neonatal periods (P0-1; P2); embryonic and early neonatal (P0-1) SMN induction robustly rescued the neuromuscular phenotype of SMA mice, while P2 induction did not [102]. Interestingly, discontinuation of SMN induction at P28 in perinatally rescued weanlings did not cause overt neuromuscular phenotypes in adulthood (one month later) [102].
These findings are supported by the work of Kariya et al. (2014), who used an inducible Cre-loxP estrogen receptor (ER) transgenic system to deplete Smn levels ubiquitously at various postnatal time points in mice homozygously expressing the SmnF7 floxed allele and SMN2 transgene; the switch from Smn requirement to insensitivity occurs abruptly at P17 [85]. This time point coincides with the end of presynaptic NMJ maturation [103] and a relative decline in the activity of Smn in assembling snRNP particles in mouse spinal cord tissue [94]. By P20, low levels of SMN (which are satisfied by two copies of SMN2 in a mouse model) adequately maintain mature neuromuscular synapses [85]. Indeed, Smn depletion had minimal consequences throughout adulthood, except for mild neuromuscular histopathology later in life, and reduced regenerative capability in response to nerve and muscle injury [85]. Thus, increased SMN dosage is required for neuromuscular maturation, as well as regeneration or repair pathways later in life. Taken together, these studies illuminate the crucial requirement for high SMN expression during the time frame of neuromuscular maturation. The need for SMN to maintain neuromuscular integrity suggests that curative therapies should be delivered during the critical stages of neuromuscular maturation in order to prevent the establishment of irreversible defects that result in lifelong neuromuscular impairment.
Our understanding of SMN temporal requirements mostly comes from mouse studies, but perhaps the most important preclinical study comes from the porcine model of SMA, as it closely resembles SMA patients. It is the only available large animal model to study SMA pathologies and therapeutics, with a size relative to human infants [104]. The pig SMA model was created by developing a shRNA that selectively knocked down porcine SMN levels to those observed in SMA spinal cord samples. The pigs developed a clear SMA phenotype at one month of age that mirrored human SMA patients and included muscle weakness with reduced electrophysiological markers of compound muscle action potential (CMAP) and motor unit number estimation (MUNE) [104]. Rescue studies to address temporal requirements of SMN were achieved by delivering scAAV9-SMN the day after injection of scAAV9-shRNA targeting pig SMN. The pigs in this presymptomatic paradigm were completely rescued; the weakness and electrophysiological parameters were corrected. When scAAV9-SMN was given at symptom onset, substantial but not complete correction was observed, and there was variation in the degree of correction when the pig had more advanced symptoms. It was found that CMAP was preserved but MUNE and motoneuron counts were not. Collectively, this study demonstrated several important points: (1) that the observed porcine SMA phenotypes were SMN dependent and could be rescued if given scAAV9-SMN presymptomatically, and (2) major phenotypic improvements and halted disease progression may still be achieved if given scAAV9-SMN early in symptomology. As expected, diminished MUNE and motoneuron loss were not rescued by postsymptomatic scAAV9-SMN treatment in these instances, since once motoneurons have been lost they cannot be replaced. Early symptomatic treatment suggests the potential for stabilization and some NMJ sprouting to occur, as observed in this study by maintenance of the CMAP [104].
6. Lower α-Motoneuron Pathologies
Development of SMA mouse models has permitted critical research on SMA disease mechanisms. In particular, mouse models have allowed for the comparison of muscles that are either vulnerable or resistant to denervation, which has provided insight into the mechanisms regulating motoneuron degeneration [86,105,106,107,108,109,110,111]. SMN deficiency within the lower motoneuron circuitry induces abnormalities in SMA model mice that ultimately result in the degeneration of vulnerable spinal motoneurons and subsequent skeletal muscle weakness [112].
6.1. Motoneuron Somas
Early in the disease process, many cellular functions and molecular signaling pathways have been shown to be dysregulated in SMN-deficient mouse spinal cord tissue and laser-captured motoneurons. Transcripts associated with translation, rRNA binding, ubiquitin homeostasis and oxidative phosphorylation are downregulated in laser-captured, preferentially vulnerable motoneurons from the Smn2B/− mouse model of intermediate SMA at a time point prior to denervation and soma loss [113]. In addition, transcripts associated with cell death are upregulated, particularly those involved in p53 signaling [113]. Compared to laser-captured motoneurons innervating relatively resistant muscles, preferentially vulnerable Smn2B/− motoneurons downregulate transcripts associated with DNA repair [113]. Similarly, intron retention, DNA damage, and p53 pathway activation are detected in spinal cord tissue 30 days postinduction of SMN depletion in antisense oligonucleotide-based inducible SMA mice [114]. Apoptosis is not observed at this time point, indicating that these changes also precede cell death [114].
A recent study by Nichterwitz et al. (2020) [115] determined that p53 pathway activation occurs in both resistant (ocular) and vulnerable (spinal) somatic motoneurons microdissected by laser capture from SMNΔ7 mice, indicating cellular stress in both populations. However, resistant (ocular) motoneurons exhibited decreased expression of pro-apoptotic genes, increased expression of survival factors, and upregulation of pathways involved in neurotransmission, neurite outgrowth, and axon regeneration. In contrast, vulnerable (spinal) motoneurons upregulated genes related to axon degeneration and axonal transport deficits [115]. Doktor et al. (2017) performed RNA sequencing on symptomatic Taiwanese SMA mice (a severe SMA mouse model) at P5, an age preceding spinal motoneuron loss [116,117]. In this study, U12-dependent intron retention was detected in all SMA tissues examined, indicative of global splicing aberrations. Gene ontology enrichment analysis revealed downregulation of angiogenesis in all tissues (spinal cord, brain, liver, and skeletal muscle), likely reflecting growth factor depletion. Additionally, differentially expressed genes in spinal cord tissue were associated with cell division and axon guidance functions [116]. Splicing defects were assessed in greater detail by Huo et al. (2014), who used microarrays to monitor splicing at exon–exon junctions and identified strong positive and negative splicing events occurring in laser-captured lumbar motoneurons from severe SMA mice (Smn−/−; SMN2+/+) at time points just preceding spinal motoneuron death (P3-P4) [118]. These findings were validated in neuroblastoma NB2a cells subjected to RNA interference-mediated Smn knockdown. Differentially spliced transcripts were associated with cellular and developmental neuronal functions, transcription, and growth control, as well as RNA metabolism [118]. Interestingly, widespread alternative splicing aberrations only arise in the late stages of the disease [119]. Taken together, transcriptional profiling of SMA mouse motoneuron and spinal cord tissues highlight early alterations in a variety of molecular pathways, including those associated with neuronal development, RNA metabolism, DNA damage, and cell death.
Concurrent with these molecular signaling changes, early functional pathologies of spinal motoneurons include hyperexcitability (hyperpolarized voltage threshold for action potential firing) [86,120,121] and reduced proprioceptive synaptic input onto spinal motoneurons [86,87,120,122]. Proprioceptive afferents sense the position of muscles in relation to the trunk [123,124] and contribute significant afferent input to modulate motoneuron firing to regulate muscle tone, which is critical for postural control [125,126]. These alterations precede spinal motoneuron loss. The molecular mechanisms evoking distinct spinal motoneuron death in SMA remain unresolved [127]. While the c-Jun N-terminal kinase (JNK)/c-jun signaling axis and the p53 signaling pathway have been implicated in severe SMA mouse models [128,129,130], the molecular mechanisms carrying out spinal motoneuron loss in SMA are likely complex, potentially context dependent, and may differ based on disease severity of the SMA model [131,132].
6.2. Motoneuron Terminals
Insight into neuromuscular pathology was largely made possible by the advent of SMA mouse models, which have demonstrated that early neuromuscular junction (NMJ) pathology is a hallmark feature of SMA. Defects at the NMJ include neurofilament accumulation in motor axons (predominantly in its phosphorylated form) [133,134,135], and fewer active zones [136,137], synaptic vesicles [77,135,136,137], and vesicle release sensors [77,138]. In addition, motor nerve terminals have fewer mitochondria [135,137,139], which is thought to be due to defects in transport [140,141] and consequently affect microtubule maturation [137]. Other key pathologies include motor end plate immaturity [26,134,135,142], neuromuscular transmission deficits [122,135,138,143,144], dysregulated calcium homeostasis [143,145], and ultimately denervation of vulnerable motoneurons [98].
Several thorough investigations of early embryonic development using SMA model mice suggest that the neuromuscular system, even in the most vulnerable muscles, undergoes relatively normal establishment [98,146,147], indicating that the deterioration of NMJs after birth occurs in the absence of major developmental disturbances [146,147]. This evidence suggests that denervation is a consequence of a failure to maintain the synapse rather than from defective axonal pathfinding and/or endplate innervation, and that defects arise during postcontact neuromuscular maturation [134]. The extent of denervation is variable but primarily distinguishable by muscle group. For example, severe denervation (>50%) appears predominantly in vulnerable axial and appendicular muscles, while other muscles are mildly or entirely resistant to denervation at end stage disease in SMNΔ7 severe SMA model mice [98]. It is important to note, however, that patterns of denervation differ amongst severe SMA mouse models; for example, Taiwanese SMA mice develop less extensive denervation than SMNΔ7 mice [98,148]. Studies investigating motoneuron/NMJ vulnerability in SMA mouse models reported no correlation between susceptibility to denervation and muscle location, muscle fiber type, nerve bundle length, NMJ size, axonal branching patterns, pruning rates, or Schwann cell expression [98,149].
7. Other Cell and Tissue Types Vulnerable to SMN Deficiency
7.1. Skeletal Muscle
Although primary deficits in SMA are caused by motoneuron-autonomous pathologies, there is mounting evidence that intrinsic muscle defects may also occur, though this phenomenon is debated. An early study by Braun et al. (1995) demonstrated that Type I and II (but not Type III) SMA patient-derived myofibers degenerate after one to three weeks in a co-culture with wild type fetal rat spinal cord explants [150], suggesting that SMN might have a muscle-specific role that is disrupted when SMN levels are exceptionally low in these cells. Supporting this hypothesis are the observations that, firstly, the Smn complex (without any associated snRNPs) localizes to the sarcomeric z-disc in striated mouse myofibrils purified from mechanically isolated myofibers [151]. However, it is important to recognize that differences in tissue preparation methods implemented prior to immunostaining for Smn in myofibrils may impact staining (e.g., using mechanically isolated myofibers then myofibril purification vs. intact muscle cryosections). Secondly, Berciano et al. (2020) demonstrated that hypertrophied (non-denervated) human Type I SMA myofibers exhibit myofibrillar ultrastructural damage and mislocalization of SMN from I-bands and M-bands to z-discs [152]. Lastly, Kim et al. (2020) have recently shown that on the background of low human SMN expression to avoid the complete absence of Smn (which is fatal), skeletal muscle-specific Smn depletion in mice (achieved using a MyoD-iCre driver to diminish SMN levels similar to that in Smn−/−; SMN2+/+ mice) induces morphological alterations to myofibers and NMJs, alters ex vivo force, impairs motor function by 6–7 months of age, and reduces lifespan [153]. However, an earlier study by Iyer et al. (2015) [154] used a myogenic factor 5 (Myf5)-Cre driver to lower SMN expression in skeletal muscle to levels similar to SMNΔ7 SMA mice, thus generating higher muscle SMN levels than those used in the Kim et al. (2020) study [153] described above. They found no muscle phenotype, weakness or reduced ex vivo force production at eight weeks of age [154]. It is unknown whether neuromuscular pathologies would have developed with increased age, as no later time points were examined. Hence, skeletal muscle cells, just like all cell other types, have an inherent SMN requirement for normal function. This issue of primary muscle dysfunction will ultimately be illuminated in the future from SMA patients identified through newborn screening whom receive nusinersen treatment that is restricted to the CNS prior to symptomology versus those whom receive SMN-inducing therapies that target the whole body (described in Section 8).
Enhanced SMN expression precedes both myoblast fusion into myotubes and motor end plate innervation, raising the question of whether SMN-deficient skeletal muscle exhibits abnormal myogenesis [155]. Several in vitro studies have demonstrated impaired myogenesis induced by SMN deficiency. When Smn levels are decreased in a mouse myoblast cell line, observed defects include reduced myoblast cell proliferation and impaired myoblast fusion into multi-nucleated myotubes [156]. Fusion deficits recapitulate observations made in myoblast cultures derived from severe SMA mouse models and from Type I SMA patients (but not from individuals with milder forms of SMA) [157,158,159].
Important experimental details should be considered when interpreting studies on intrinsic muscle defects in SMA. These include species-specific differences between humans and mice, which likely include discrepancies in typical/required SMN dosage, as well as the specific SMA mouse models utilized (as each express different SMN dosages most likely due to position effects of transgene integration sites and/or promoters used). This leads to differences in SMA disease severity of functionally Smn-null mice homozygously expressing the transgenic SMN2 allele. An example of this discrepancy is the transgenic SMN2 line 89 (TgSMN2-Ahmb89), which is driven by the human SMN2 promoter sequence and results in a very severe phenotype [111], versus a transgenic SMN2 allele, which is under the control of murine Smn promoter and results in a mild phenotype [160].
7.2. Schwann Cells
A loss of non-myelinating Schwann cells may also influence neuromuscular pathology. Perisynaptic Schwann cells are reduced in number in SMA model mice in vulnerable and resistant muscles [136], fail to completely cover endplate sites [161], and express fewer key proteins required to generate the peripheral extracellular matrix [162,163]. Furthermore, selective restoration of SMN in Schwann cells improves neuromuscular function [163].
7.3. Astrocytes
In addition to muscles and motoneurons, SMN deficiency affects astrocytic function, which likely influences SMA pathogenesis in severe cases. Postmortem analysis of SMA patient spinal cord tissue reveals astrogliosis [164], and cultured astrocytes differentiated from SMA patient-derived iPSCs exhibit morphological and functional alterations consistent with astrocytic activation [165]. Reactive astrocytes were also observed in SMNΔ7 mouse spinal cords at ages preceding spinal motoneuron loss [165]. Critically, astrocyte-specific SMN repletion attenuates denervation, partially mitigates stripping of proprioceptive synapses onto spinal motoneurons and greatly enhances survival of Smn2B/− mice; repletion of SMN in SMNΔ7 mice also enhances survival, although this benefit was moderate [164]. Additionally, abnormal calcium regulation and reduced growth factor production has been observed in SMN-deficient astrocytes [165]. Overall, an emerging body of work indicates that low SMN levels in astrocytes located in the spinal cord may contribute to SMA disease onset and/or progression.
7.4. Heart
Low SMN levels may induce dysfunction in cell types beyond the neuromuscular system. In exceptional cases, which comprise infants with the most severe form of SMA (Type 0), defects in fetal cardiac development have been reported [25]. The most common abnormality is septal and cardiac outflow tract defects, which may contribute to rare but reported distal necrosis [166,167]. Additionally, benign cardiac arrhythmias have been reported in patients with milder forms of SMA [168], although these rhythmic abnormalities may be a consequence of physical inactivity and trunk muscle weakness [169]. Although anatomical and functional cardiac defects are not often observed in SMA patients, they are pervasive in severe SMA mouse models [106,170,171,172,173,174,175,176,177,178,179].
7.5. Additional Susceptible Cell and Tissue Types
A variable degree of thalamic dysfunction [180] and thalamic neuronal degeneration and gliosis has also been reported in severe SMA patients at end stage of the disorder [26]. Additionally, other abnormalities have been reported, including abnormalities in autonomic, sensory, gastrointestinal, and endocrine systems [22,24,26,181,182,183,184]. In animal models, a number of organ phenotypes have been noted, including abnormalities in cardiac, lymphatic, kidney, liver, pancreas, spleen, vasculature, bone and connective tissues (thoroughly reviewed in Yeo and Darras (2020) [185]). These non-canonical pathologies are generally only reported in patients with the most severe forms of SMA, suggesting that even low expression levels of SMN (achieved by 2–3 functional copies of SMN2) is sufficient for vitality in these tissues.
Use of SMN-based genetic therapies in humans may uncover pathology in non-motor systems, particularly in severe SMA patients who would otherwise experience a robust motor phenotype and gravely shortened lifespan that could obfuscate other organ impairments not readily apparent during the natural disease progression. Now that disease-modifying therapies are available to treat primary SMA pathologies (e.g., spinal motoneuron dysfunction and loss), secondary defects arising from chronic SMN deficiency in untargeted peripheral tissues may emerge. Future clinical research that follows Type I patients receiving FDA-approved therapies that target the central nervous system (via antisense oligonucleotides) versus the whole body (by gene therapy or oral small molecule) will shed light on the importance of SMN-deficient peripheral cell types in human SMA.
8. The Quest for Effective SMA Therapies: FDA-Approved SMN-Dependent Therapeutics
The last decade of preclinical research searching for SMA treatments has resulted in significant advancements in our understanding of the biologic, cellular, and genetic mechanisms underlying SMA. Despite this knowledge, several challenges have made drug development difficult. SMA comprises a broad spectrum of phenotypes, with a large population developing an onset of symptoms during infancy. Additionally, therapeutic treatment must be able to effectively target disease-relevant tissue (such as lower α-motoneuron somata in the central nervous system and NMJs in the peripheral nervous system). Genotype/phenotype studies in humans and preclinical investigations have shown that the best therapeutic approach to preventing or improving disease progression is through increasing functional SMN levels. Animal model studies suggest that even a relatively modest increase in SMN, when given early enough, produces clinically meaningful improvements [104,186,187,188,189]. Notably, despite early intervention being crucial for optimal improvement, animal models suggest that restoration of SMN later in life may still provide some therapeutic benefit [95,104]. Notwithstanding these temporal and cell type-specific challenges, the first FDA-approved, SMN-based treatment became available in December 2016, and in the four years since this approval, several other promising candidates and two more FDA-approved therapies have followed (Figure 4).
Several SMN-based therapeutic approaches have been investigated to upregulate functional SMN protein. Examples of therapeutic approaches include targeting SMN2 splicing (antisense oligonucleotides and other small molecules), transcription (histone deacetylase inhibitors, hydroxyurea, lncRNA-targeting oligonucleotides, prolactin, and quinazoline), translation (Indoprofen, aminoglycosides), as well as stabilization of SMN transcript or protein (Celecoxib) and the insertion of SMN genes (adeno-associated viral and lentiviral vectors) [190,191]. Currently, three SMA-modifying therapies have been approved by the FDA (detailed in the following sections), and thus far, all SMN-based approaches have similarly altered the disorder progression and outcome of SMA-affected individuals. These types of therapies are currently the best method to prevent motoneuron degeneration (if administered early enough); however, they may not be entirely curative for all patients receiving treatment. While most patients who receive presymptomatic SMN-based treatment remarkably achieve motor skills in the normal developmental range [192], there remains a population of SMA-affected individuals who would benefit from additional treatments to address persistent dysfunction. This population includes patients who have one SMN2 copy, patients with two SMN2 copies but experience suboptimal motor development, or patients who receive postsymptomatic treatment (when substantial motoneuron death has already occurred). These individuals will likely require additional treatment strategies to improve residual motor dysfunction.
8.1. Nusinersen: An Antisense Oligonucleotide (ASO)
The endogenous presence of a paralog gene that produces the necessary protein has made SMN2 an ideal therapeutic target. If splicing of SMN2 is corrected to produce a greater percentage of full-length SMN protein (similar to levels produced by SMN1), symptoms of SMA can be alleviated. The first FDA-approved therapy for SMA uses an ASO under the generic name nusinersen (brand name Spinraza®). Nusinersen uses a synthetic strand of nucleic acids linked together with a 2′O-methoxyethyl backbone that functions by recognizing and binding to cellular RNA to correct gene splicing. Nusinersen uses Watson–Crick pairing to specifically bind the intronic splicing silencer-N1 (ISS-N1) sequence in SMN2, which is a major inhibitory element regulating the splicing of exon 7. ISS-N1 has proven to be a model target for ASOs to increase the ratio of full-length SMN protein derived from SMN2 transcripts, as ISS-N1 inhibition results in the inclusion of exon 7 in mature SMN2 transcripts [193,194].
ASOs are endocytosed by cells upon intrathecal administration, though the mechanisms regulating absorption are poorly understood. Upon entrance to the cell body, nusinersen ASOs enter the nucleus where they bind to SMN2 pre-mRNA transcripts to correct exon 7 splicing and thus increase full-length SMN levels (Figure 4B). Nusinersen has drastically altered the outcome and progression of this previously unmodifiable neuromuscular disorder [195], and current efforts are underway to improve ASO penetration, durability, and therapeutic safety and efficacy profiles.
Preclinical studies in mice have shown that administration of ASOs targeting ISS-N1 is sufficient to mitigate neuromuscular pathology and greatly improve survival and motor function in a time- and dose-dependent manner [173,188,189,196,197,198]. In non-human primates, a single intrathecal bolus injection of nusinersen was observed to be widely distributed throughout the spinal cord, and predominantly accumulated in small and large cell bodies of the grey matter (consistent with neural and glial cell targeting) [199]. When administered to Taiwanese Type III SMA (Smn−/−; SMN2+/+) mice, ASO-induced SMN2 splicing changes were still detectable after six months [199], suggesting that the long half-life of the drug permits several months between treatments for SMA patients [200]. Indeed, patient autopsy results demonstrate that intrathecal delivery of nusinersen elevates SMN2 mRNA exon 7 inclusion in neurons and other cell types in the spinal cord [201].
Clinical studies of nusinersen in all types of SMA patients showed encouraging efficacy, tolerability, and pharmacology consistent with its intended mechanism of action. Intrathecal injections were well tolerated and improved motor function in a majority of treated patients [44,46,200]. Type I SMA patients had remarkably improved lifespans compared to what would have been normally expected from the natural history of the disorder, and patients experienced fewer respiratory complications requiring ventilation [46]. More historical data are necessary to determine lifespan benefits of individuals with less severe forms of SMA. Type I SMA patients receiving nusinersen therapy achieved motor milestones that are unprecedented in the natural disorder progression, and many achieved these skills within normal time frames of motor development [46].
Patients with later-onset SMA that have received nusinersen therapy report a better quality of life, and caregivers of these patients also report a decreased impact of caring burden in addition to a greater quality of life [202]. Adult patients receiving nusinersen therapy experienced a stabilization of motor function or a reduction in symptom severity, though this improvement was not observed in all patients [203,204]. In further support of the limited benefit of delayed nusinersen treatment, Arnold et al. (2016) demonstrated that neuromuscular deficits persist in SMNΔ7 mice treated with ASOs at P4-6 [205]. The persistence of motor impairment suggests that neuromuscular function is a promising target for additive pharmacological interventions, particularly for patients that receive SMN-based treatment during adulthood.
The above treatment trials outline the effect of nusinersen treatment postsymptomatically. An interim report of the NURTURE study, an ongoing, open-label, multi-site single-arm, Phase 2 trial, which enrolled and treated SMA patients in infancy while they were presymptomatic, illustrates the importance of proactive treatment with nusinersen as soon as possible after establishing an SMA genetic diagnosis [192]. Infants enrolled in NURTURE were ≤6 weeks of age at first dose and, based on SMN2 copy number and expected concordance of phenotype with an affected sibling(s), were predicted to have type I or II SMA (15 children with 2 SMN2 copies and 10 children with 3 SMN2 copies). At a median 2.9 yrs of follow up, all patients were alive without requirement of permanent ventilation, which directly opposes the natural history of untreated Type I SMA infants. Motor function was assessed using several assays, including crawling, sitting, standing, and walking (with or without assistance). All 3-copy SMN2 patients (10/10) achieved motor milestones in timelines consistent with normal development, and all patients achieved independent sitting. A majority of 2-copy SMN2 patients achieved motor skills unexpected in the natural history of the disease, though more difficult tasks, such as standing or walking alone, were not achieved by all children in this interim report (3/15, or 20%). However, the remaining 80% of 2-copy SMN2 patients were able to stand or walk, though only 27–40% of patients (4/12 stand alone, and 6/15 walk alone) achieved this during normal developmental time frames. [192]. Essentially, all of the study trial result outcomes: (1) exceeded those of participants’ SMA-affected siblings who had not received this early treatment; (2) exceeded expectations based on the natural history of SMA; and (3) treatment benefits exceeded those observed when treatment is initiated in the symptomatic period. Additionally, on a molecular level, phosphorylated neurofilament heavy chain (p-NF-H) levels have been shown to be a potential biomarker of disease severity and treatment response in SMA patients who received nusinersen treatment symptomatically [192,206,207]. pNF-H is a neuron-specific cytoskeletal structural protein that is released into the CSF and plasma when axonal damage occurs [208]. The presymptomatic NURTURE trial demonstrated increased pNF-H levels at baseline patient identification [192], indicating that the disease is biologically active, warranting early treatment. Like the other trials [206,207], pNF-H levels rapidly declined postnusinersen treatment and then stabilized to a lower plateau level [192]. Collectively, these results highlight the need for early population-based newborn screening for SMA and initiation of treatment as soon as possible thereafter with SMN-inducing therapies. Furthermore, these results suggest that some 2-copy SMN2 patients (particularly those who struggle to achieve the ability to stand or walk unassisted) would benefit from an additional, SMN-independent therapy that specifically targets neuromuscular function.
Despite immense benefits that alter the natural history of SMA, the use of nusinersen for the treatment of SMA is not without challenges. Intrathecal administration is relatively invasive, requiring sedation or anesthesia, and can be particularly problematic in young patients or patients with scoliosis. Since ASOs have a limited penetrability of the blood–brain barrier, direct administration into the cerebrospinal fluid is required to achieve endocytosis into motoneuron somata. Additionally, intrathecally administered ASOs have poor peripheral penetrance [209]; the clinical implications of which (if any) remain to be determined as nusinersen-treated individuals continue to age. Potential risks of treatment with nusinersen are thrombocytopenia and coagulation abnormalities, as well as renal toxicity [210].
In the United States, nusinersen is currently approved for the treatment of any patient with biallelic mutations or deletions of SMN1 [211]. During the first year of treatment, nusinersen is estimated to cost $750,000 for the six recommended doses. Thereafter, the medication costs $375,000 for the recommended three maintenance doses per year. No cessation of annual treatments is recommended per se, unless the patients (or their caregivers) elect not to continue treatment.
8.2. Onasemnogene Abeparvovec-Xioi: A Self-Complimentary Adeno-Associated Virus (scAAV9)
The second approved therapy for SMA utilizes recombinant adeno-associated viral vectors to target the central underlying deficiency that causes SMA (Figure 4E). Onasemnogene abeparvovec-xioi (previously known as AVXS-101, and now as the brand name Zolgensma®, and hereafter referred to as onasemnogene-xioi) is available to patients under 2 years of age (and not exceeding 13.5 kg) with biallelic mutations or deletions of SMN1. Onasemnogene-xioi is a self-complementary adeno-associated viral vector serotype 9 (AAV-9) carrying human full-length SMN cDNA under control of the hybrid CMV enhancer/chicken β-actin promoter. Upon intravenous delivery, the non-replicating scAAV9 crosses the blood–brain barrier into the central nervous system, where it is endocytosed by cells, including motoneurons, and trafficked to the nucleus. Once in the nucleus, the virus uncoats and transduces the host cell to transcribe its double-stranded DNA unit (full-length SMN cDNA) [212]. This medication requires a single intravenous administration over a period of 60 min to achieve therapeutic benefit.
Targeting of cells within the central nervous system is achieved by the use of the AAV9 serotype, which has been shown to efficiently transduce neurons and glia in rodents, pigs, and non-human primates [104,187,213,214,215]. However, transduction efficiency is age dependent and varies between species and cell types. Studies in neonatal mice have demonstrated that peripheral administration of GFP-tagged scAAV9 with a chicken β-actin hybrid promoter transduced 60% of motoneurons, and was measurable 20 days after intravenous injection [215]. Using this vector for SMN gene replacement in SMNΔ7 neonates rescues motor function, neuromuscular physiology, and survival, while intravenous administration to P10 SMNΔ7 mice yields limited benefits [187], supporting the established crucial need for early intervention. In addition to disease progression prior to treatment, this result is also attributed to an age-related switch in transduction efficiency from neurons to glia in mice [215]. This age-dependent switch in transduction efficacy does not appear to occur in larger mammals [104,216]. Using a novel porcine model of SMA, Duque et al. (2015) elegantly demonstrated that intrathecal delivery of scAAV9-SMN corrected SMA phenotypes when given early (presymptomatically), but was not completely curative when delivered at symptom onset (but it did halt disease progression), due to motoneuron loss that had already occurred prior to treatment [104]. When scAAV9-GFP was tested in newborn (P0–P90) and 3-year-old cynomolgus macaques, systemically administered scAAV9 crossed the blood–brain barrier and robustly transduced spinal motoneurons, brain cells (in particular, glial cells), and skeletal muscle (though more so in the young animals) [213]. 3-year-old macaques had less frequent GFP+ expression in spinal motoneurons compared to newborns [213], although possibly due to a dosing difference. Collectively, these studies suggest that through an intravenous route, onasemnogene-xioi administration to SMA patients may have restricted benefits to phenotype when administered postsymptomatically, primarily due to inability to recover motoneurons that have already been lost.
Clinical trials of onasemnogene-xioi have demonstrated a remarkable improvement of motor function in SMA patients when treated presymptomatically. Patients with three copies of SMN2 achieved motor skills within normal age ranges of acquisition. In comparison, a majority (but not all) of patients with two copies of SMN2 also achieved motor milestones within normal ranges. A long-term follow up of patients who were treated postsymptomatically showed that no patients lost any achieved motor skills, which is a stark contrast to the natural history of this disorder. The most commonly achieved motor milestone included head control, rolling from back to side, and sitting unsupported for more than 30 s. However, less than half of the patients receiving postsymptomatic intervention achieved advanced motor skills such as walking or standing (supported or unsupported) [217]. Additionally, onasemnogene-xioi dramatically improved respiratory function regardless of pre or postsymptomatic administration. In comparison, over 90% of patients with severe forms of SMA not receiving SMN-based therapy require permanent ventilation by their second birthday.
While gene therapy has remarkably changed the field of SMA treatment, use of onasemnogene-xioi is not without potential drawbacks. The inherited prevalence of maternally-derived (though potentially transient) neutralizing antibodies to AAV9 may restrict the use of this type of therapy in a minority of SMA-affected individuals due to the risk of rendering the viral vector ineffective [218,219]. Furthermore, gene transfer does not permit dose cessation if a safety issue arises. In particular, onasemnogene-xioi has a potential risk of serious liver complications, and requires monitoring of liver function prior to and for at least 3 months after infusion [220]. Thrombocytopenia and elevated troponin-I are also potential risks, requiring platelet counts and troponin-I levels to be monitored before and after treatment [220]. Additionally, unlike antisense oligonucleotides that target the endogenous SMN2 gene(s), AAV9 vectors do not have an inherent ceiling of SMN upregulation. A final consideration should also be given to the potentially prohibitive cost of onasemnogene-xioi—at $2.1 million for a single dose, this medication is currently (January 2021) the world’s most expensive drug [221].
8.3. Risdiplam: An Oral, Brain-Penetrant Small Molecule
The most recently approved therapy for SMA is EvrysdiTM (risdiplam), which is a daily, orally bioavailable small molecule that is brain penetrant. Similar to nusinersen, risdiplam is an SMN2 exon 7 splice modifier (Figure 4C). This molecule promotes exon 7 inclusion by binding to two sites in SMN2 pre-mRNA: the 5′ splice site (5′ss) of intron 7 and the exonic splicing enhancer 2 (ESE2) of exon 7. This binding stabilizes a ribonucleoprotein (RNP) complex that is critical to the specificity of this small molecule for SMN2 over other genes [222]. Risdiplam has been approved by the FDA for all SMA patients 2 months or older, and clinical trial results show significant improvement in motor function and SMN levels, with the most notable benefits in younger patients [223,224,225]. Approximately 90% of Type I SMA patients were alive after 12 months of treatment (and reached 15 months of age or older), and none required permanent ventilation at 28 months of age [226]. Remarkably, 41% of treated Type I SMA infants achieved unsupported sitting for 5+ s [226]. Type II–III SMA patients also experienced significant and sustained improvements in motor function compared to placebo-treated controls after 12 months of risdiplam treatment [226].
Risdiplam has a list price of $340,000 per year for patients weighing 20+ kg, with the price adjusted for patients under this weight limit. Risdiplam is an attractive competitor to nusinersen and onasemnogene-xioi not just due to its lower price but also because of the convenience of at-home administration, as risdiplam can be swallowed as a flavored liquid or given in a feeding tube. Since a large percentage of SMA patients (particularly Type II SMA) experience severe scoliosis (which can complicate or deter intrathecal administration), patients, families, and health care providers may favor the use of an orally administered drug. Additionally, this method of administration permits risdiplam to be bioavailable in peripheral systems, in comparison to the limited ability of centrally administered treatments to reach peripheral organs [227]. Bioavailability in the periphery may mitigate emergence of potential secondary defects arising from SMN deficiency in peripheral tissues. More clinical research is necessary to compare the short- and long-term tolerability, safety, effectiveness, and peripheral distribution of risdiplam, nusinersen and onasemnogene-xioi therapies.
8.4. Cost–Benefit Considerations for Approved SMN-Based Therapies
While many American insurance companies have decided to cover SMN-based therapies, some companies have implemented stricter guidelines for patient eligibility than recommended by the FDA, such as age limitations or the requirement of symptom manifestation prior to treatment coverage [228]. These company-imposed restrictions are driven by the high drug price tag. Additionally, outside the United States, widespread approval for use of nusinersen as a therapy for SMA has been slow, primarily hampered by the high drug price [229].
For example, a recent survey indicated that insurance approval is a critical barrier to accessing nusinersen, especially among adult patients [230], as ~25% of respondents declare insurance ineligibility as one the reasons for not currently receiving nusinersen treatment. This self-reported barrier supports the notion that insurance-dictated eligibility can limit or delay treatment. In particular, adults with SMA may have already experienced significant motoneuron death, and additional delays in treatment may severely restrict any therapeutic benefit of SMN-based therapy.
The high cost of these drugs does not include the additional and substantial medical costs associated with treatment (including but not restricted to medical appointments and follow ups, administration costs, income loss due to illness or time off work, and other “invisible” costs incurred by caregivers). One survey reports that, on average, patients and/or caregivers drive 3.52 h (2 h median) to nusinersen administration centers [230], which demonstrates that significant resources (such as time and/or vehicle access requirements) are required to pursue treatment. These “invisible” costs are important to consider for health care providers and SMA-affected individuals (including caregivers) when deciding which treatment(s) to pursue.
Comparing treatment costs across patient populations is not straightforward, as two factors can influence the overall cost of medication. First, differences in dosing (single versus multiple) will dictate medical cost, though this cost may be difficult to determine as there is no recommended cessation of either risdiplam or nusinersen. Second, overall cost is also dependent on when the individual starts receiving treatment. Despite these complications in determining cost–benefit outcomes, some analyses have been made assessing the value of nusinersen and onasemnogene-xioi. A report released by the Institute for Clinical and Economic Review comparing the quality-adjusted life-year (which compares drug price tag to benefits in disease burden, assessing both quality and quantity of life) suggests that the nusinersen list price should be discounted 10-fold, and the onasemnogene-xioi list price should be discounted 2-fold [231,232]. At the time of these studies, risdiplam had yet to be approved by the FDA and was thus not evaluated. Now, with risdiplam’s FDA approval and its lower pricing, this drug may competitively drive drug costs lower for nusinersen, onasemnogene-xioi, and future disease-modifying therapies.
9. The Quest for Additional SMA Therapies: SMN-Independent Therapeutics
SMN-based therapies remain at the forefront for SMA treatment strategies, but not all patients receiving SMN-dependent medicine experience full symptomatic relief. Furthermore, not all individuals with biallelic SMN1 mutations/deletions are eligible for insurance coverage of genetically targeted therapies. The risk of potentially serious adverse side effects has resulted in a debate about treating presymptomatic infants with four or more copies of SMN2 [233,234]. However, individuals with four copies may still develop motor dysfunction [235] and thus would benefit from SMN-based therapies. Nonetheless, complex treatment decisions require parents to balance potential risks, cost, and accessibility. Additionally, insurance companies may restrict eligibility for costly SMN-based treatments, which may impact the decision of whether to treat presymptomatic individuals with 4 SMN2 copies. Another SMA population that has been questioned for eligibility of SMN-based treatment(s) is adults that have already experienced significant motoneuron loss, who may not extensively benefit from SMN-based therapeutic approaches. For these reasons, an extensive number of SMN-independent therapeutic strategies have been investigated, which target a wide range of affected cells to protect and improve function. Ideally, these therapeutic strategies will be available to patients across the SMA spectrum and used in tandem with SMN-dependent approaches.
9.1. Neuroprotective Strategies
Neuroprotective strategies encompass SMN-independent treatments that are targeted to prevent dysfunction in motoneurons and associated circuitry. While several neuroprotective treatments have been previously attempted in other neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS), relatively poor outcomes of clinical trials have limited widespread investigation of neuroprotective agents for SMA. Neurodegeneration is a complex, multi-cellular process and, consequently, targeting single cell pathways contributing to death or survival may not be sufficient to halt or improve disease progression. Therefore, any employed neuroprotective strategies would likely require complementary SMN-targeted treatments to derive benefits.
There are two neuroprotective agents that have been approved for use in other diseases that have been tested for efficacy in SMA patients. Gabapentin, an anticonvulsant used to treat neuropathic pain and restless leg syndrome, has been evaluated due to its ability to decrease glutamate signaling and thus decrease excitotoxicity. However, two large clinical trials composed of Type II–III SMA patients demonstrated minimal effects on motor function [49,236,237]. Another drug, riluzole, has been used to treat ALS patients with some benefits. ALS patients receiving this drug have lifespans extended by 2–3 months, though with no benefit to motoneuron function. Use of riluzole in SMA animal models appeared promising, where it was able to improve neuromuscular defects in a C. elegans model [238], and lifespan and cytoskeletal organization in a mouse model [239]. However, a small clinical trial comprising 10 Type I SMA infants (7 riluzole-treated, 3 placebo-treated) was cut short due to lack of improvement in motor milestones, though the small cohort underpowered statistical assessment. A second clinical trial (Clinicaltrials.gov: NCT00774423) evaluated the effects of 50 mg of riluzole in Type II–III SMA patients, which has been shown to be a sufficient dose for pharmacokinetic exposure in younger SMA patients [240]. However, results of this trial have yet to be published (as of December 2020).
One neuroprotective approach that has been evaluated in SMA animal models utilizes enhancers or mimetics of neurotrophic factors. More than a dozen endogenous trophic factors have been discovered to modulate motoneuron survival in vitro, including but not limited to brain-derived neurotrophic factor (BDNF), neurotrophin-3, vascular endothelial growth factor, glial cell-derived growth factor (GDNF), ciliary neurotrophic factor (CDNF), agrin, and insulin-like growth factor-1 (IGF-1). Historically, BDNF, GDNF, and CDNF have been extensively investigated to prevent motoneuron degeneration and improve motor function [241,242,243,244,245,246,247]. However, much of the work in the SMA field has focused on IGF-1, BDNF, and agrin.
Low levels of IGF-1 have been detected in severe SMA mouse models, which are restored to normal levels upon correction of SMN levels [173,248]. Overexpression or mimetic administration of IGF-1 to SMNΔ7 mice improves lumbar motoneuron degeneration, cardiac defects, skeletal myofiber size, and motor function, but has limited benefit to survival [179,248,249,250]. Administration of AAV-driven IGF-1 into deep cerebellar nuclei of intermediate SMA model mice improves motoneuron degeneration but has no effect on neuromuscular pathology [251]. These studies indicate that IGF-1 may be a potential target for neuroprotective approaches, but is likely to have restricted benefits if used without complementary treatment(s).
The effects of BDNF have also been explored in developing motoneuron cultures and SMA models. Through its mechanism of action on TrkB receptors, BDNF application augments calcium transients via increased Cav2.2 clustering, and improves F-actin assembly and growth cone formation in motoneurons in vitro [252], suggesting that some of the developmental defects seen in SMA model cultured motoneurons [145] and mice [253] are consequences of reduced BDNF-mediated trophic support. SMN-regulated BDNF expression has also been explored in SMA model NSC-34 motoneuron-like cells. Smn deficiency results in the downregulation of the Akt signaling pathway [254], which is regulated downstream by BDNF binding to TrkB [255]. Application of loganin (a neuroprotective iridoid glycoside) to NSC-34 cells increased neurite length, cell viability, and Smn expression, upregulated BDNF and activated the Akt pathway in these cells. Loganin administration to SMNΔ7 mice improved motor function and mildly improved lifespan, and blockade of the IGF-1 receptor attenuated the protective effects of loganin [254].
The neural form of agrin (z+ agrin) is another trophic factor that is significantly reduced in SMA model mice [256]. Administration of NT-1654, a cleavage-resistant splice variant of z+ agrin with synaptogenic properties, significantly improved motor function and neuromuscular pathology [257]. The mechanism(s) regulating the reduction in trophic factors in SMN-deficient systems requires additional investigation, but nevertheless may be therapeutic targets to improve the integrity of motor circuits.
Finally, the neuroprotective agent olesoxime, which has been shown to promote neuronal survival in cultured cells deprived of trophic factors [258], demonstrated no significant benefit to Type II–III SMA patients in the OLEOS clinical trials [259]. This further supports the conclusion that neuroprotective strategies are unlikely to ameliorate motor dysfunction when used alone, and thus require a combinational approach to maximize therapeutic benefit.
9.2. Muscle-Directed Strategies
Given the intrinsic role of SMN in neuromuscular development, strategies that enhance muscle function have been extensively explored. One of the most investigated targets with promising results in milder SMA mouse models or as a complementary treatment to severe SMA mouse models receiving SMN-based therapy has been the inhibition of myostatin. Myostatin is a member of the transforming growth factor Β superfamily, and is predominantly synthesized and expressed in skeletal muscle. Myostatin acts as an endogenous negative regulator of skeletal muscle growth and size [260,261,262], and is neutralized by the autocrine glycoprotein follistatin and myostatin propeptide [263]. Studies evaluating the benefits of recombinant follistatin administration to SMNΔ7 mice observed improvements in lifespan, motor function, and motoneuron death [264]. However, studies directly inactivating myostatin in SMNΔ7 mice did not significantly ameliorate motor function, fiber size, or survival [265,266], likely due to the rapid disease progression in this mouse model. Nonetheless, myostatin inhibition may still provide therapeutic benefit in milder forms of the disorder and when provided in combination with SMN-based genetic therapies: one study evaluating AAV-driven myostatin inhibition in SMNΔ7 mice treated with SMN-based ASOs observed improvements in weight gain, motor function and endurance, survival, proprioceptive synapses onto motoneurons, as well as mitigated neuromuscular pathology [267]. Another study evaluating myostatin inhibition in mild SMA model mice (generated by treatment with a suboptimal dose of an SMN2-splicing modifier) demonstrated efficacy in late disease stages [268]. Myostatin is the therapeutic target for SRK-015, which is a monoclonal antibody that blocks the activation of the latent form of myostatin rather than inhibiting the mature myostatin form or blocking its receptor [269]. Preclinical studies in a pharmacologically induced SMA mouse model have shown that both early and late administration of muSRK-015P (a suboptimal variant of SRK-015) increases muscle mass and motor function [270]. A clinical trial is underway assessing the benefits of intravenously administered SRK-015 for Type II–III patients (Clinicaltrials.gov, NCT03921528), primarily for use as a complement to SMN-based therapy. This treatment is one of the first muscle-directed therapies to improve muscle atrophy in SMA mouse models of varying disease severity.
Another muscle-centric approach utilizes fast skeletal muscle troponin activators. Troponin complexes regulate contraction in skeletal and cardiac muscles. Fast skeletal troponin activators increase calcium affinity of the troponin-tropomyosin regulatory complex, which results in the sensitization of the sarcomere to calcium concentrations and improves skeletal muscle contractility and muscle performance. Force–calcium and force–frequency relationships are shifted leftward by troponin activation. Thus, increased contractility is observed even with inadequate neural signaling because these activators amplify the response of the muscle to submaximal nerve stimulation [271]. Reldesemtiv (CK-2127107), a second-generation fast skeletal troponin activator, has been shown to increase force production by the tibialis anterior upon transcutaneous deep fibular nerve stimulation in healthy adults [272]. Combinatorial treatment of Taiwanese SMA model mice with SMN-based genetic therapeutics and reldesemtiv enhanced the force produced by in vivo plantar flexion more than either SMN upregulator alone [273,274]; however, these preclinical data are currently unpublished. Clinical trials for Reldesemtiv have completed phase 2 of clinical trials (Clinicaltrials.gov, NCT02644668), but a literature summary of results have not yet been published (as of December 2020).
9.3. Drugs Targeting Neuromuscular Function
Despite our superficial understanding of the mechanisms regulating NMJ breakdown, a large and reproducible body of evidence supports the notion that neuromuscular junctions undoubtedly contribute to the disease pathogenesis in SMA. Reduced neuromuscular transmission and increased NMJ failure is characteristic of the SMA phenotype and has led to the testing of drugs that improve neuromuscular communication. However, few drugs have provided symptomatic relief for SMA patients. Pyridostigmine, an acetylcholinesterase inhibiter canonically prescribed to patients with myasthenia gravis, was reported to increase stamina in 2 of 4 Type II–III SMA patients [275]. Though limited, this positive finding supported the evaluation of pyridostigmine in a clinical trial comprised of patients with Type II–IV SMA living in the Netherlands (Clinicaltrials.gov: NCT02941328). While the trial was completed in 2018, results have yet to be published (as of January 2021).
Similarly, the voltage-gated potassium channel antagonist 3,4-diaminopyridine (3,4-DAP) is also undergoing two clinical trials for patients with Type III SMA (Clinicaltrials.gov: NCT03781479, NCT03819660). 3,4-DAP is canonically prescribed to patients with Lambert-Eaton myasthenic syndrome (LEMS), which is a neuromuscular disorder similarly characterized by a reduction in presynaptic calcium influx and reduced neurotransmitter release. However, dose-dependent side effects of 3,4-DAP (caused by blood–brain barrier penetration) limit symptomatic relief in patients with LEMS, so it is possible that patients with SMA will similarly experience limited benefits with 3,4-DAP. Another similar voltage-gated potassium channel-blocking drug being tested in clinical trials is 4-aminopyridine (4-AP; Clinicaltrials.gov: NCT01645787). 4-AP has traditionally been used to treat patients with multiple sclerosis due to its more efficacious penetrance of the blood–brain barrier compared to 3,4-DAP; however, similar to 3,4-DAP, 4-AP has dose-dependent side effects that restrict the use of an optimal dosage for motoneuron function. Results from these clinical trials have also not yet been published [49]. Similar to other SMN-independent approaches discussed in this review, these neuromuscular-targeted medications are unlikely to immensely benefit SMA patients when used as a stand-alone therapy. In particular, these drug interventions occur after the critical period of neurotransmission-regulated NMJ development, and thus are unlikely to reverse established neuromuscular defects [276]. However, if neuromuscular-targeted medicines are utilized during the critical period of NMJ development, they may provide long-term and substantial therapeutic benefits, as described below.
One recent investigation by Tejero et al. (2020) [253] investigated the effect of (R)-Roscovitine, a cdk-5 inhibitor with positive allosteric effects on voltage-gated calcium channels (Cav2.1-Cav2.2), on SMA pathology during motoneuron development. Application of (R)-Roscovitine to SMA motoneuron cultures increased Cav2.2 channel clustering, spontaneous calcium transients, elongated axons, and improved neurotransmission [253]. Axonal elongation was similar when motoneuron cultures were exposed to a derivative of (R)-Roscovitine, GV-58, which is a molecule with more potent Cav2.1-2.2 effects and no significant cdk activity at physiological ATP levels [277]. Additionally, systemic administration of (R)-Roscovitine to pregnant dams (E11.5-17.5) significantly increased the lifespan of SMNΔ7 mice. This lifespan benefit was independent of effects on cdks, as (S)-Roscovitine (which lacks Cav2 activity but retains cdk-5 inhibition) did not improve lifespan [253].
Another preclinical study evaluated the effect of GV-58 alone, or in combination with 3,4-DAP, which has been shown to synergistically increase neuromuscular transmission in Lambert Eaton myasthenic syndrome-affected NMJs [278]. The use of both GV-58 with 3,4-DAP restored neuromuscular transmission to control levels in ex vivo neuromuscular junctions in SMNΔ7 mice, but GV-58 alone was sufficient if the mice had been treated with ISS-N1 ASOs at birth. Similar results were observed in vivo, when measuring changes in muscle strength after an acute subcutaneous administration of GV-58 and 3,4-DAP to P10 SMNΔ7 pups. Untreated SMNΔ7 mice maximally benefited from the combination of GV-58 and 3,4-DAP, while ASO-treated SMNΔ7 mice maximally benefitted from GV-58 alone (with no additive benefit of 3,4-DAP) [279,280]. Results from these studies suggest that targeting calcium homeostasis in developing motor nerve terminals may remarkably alter neuromuscular dysfunction and enhance motor ability, and can be used to complement SMN-dependent approaches.
9.4. Endogenous SMN-Independent Protective Modifiers
An attractive target for additive therapies includes endogenous disease modifiers, but the last decade of research has produced controversial and perplexing observations. Family studies indicate that the SMN2 gene is the main modifier of the SMA phenotype, where more SMN2 copies equate to a milder phenotype, but this observation is not absolute, and other modifiers exist both within and outside of the SMN2 region [9,11,12,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296]. Transcriptome-wide differential expression analysis of genes from SMA-discordant families have elucidated endogenous protective modifiers of the SMA phenotype in some individuals, but these modifiers are not ubiquitously associated with phenotypic improvement across all SMA patients [282,294,297] Curiously, several of these putative modifiers function by sensing or regulating calcium. These positive modifiers include plastin 3 (PLS3) [80,290,298], neurocalcin delta (NCALD) [299], and calcineurin-like EF-hand protein 1 (CHP1) [300].
Some have proposed that the downregulation of the neurocalcin delta (NCALD) gene is a protective modifier of the SMA phenotype. NCALD functions as a neuronal calcium sensor to negatively regulate endocytosis. Heterozygous knockdown of NCALD improves endocytosis in fibroblasts derived from SMA patients. Knockdown also improves axon elongation and NMJ size in severe and mild SMA model mice, and accelerates neuromuscular maturation and improves motor function in intermediate SMA model mice. Additionally, suppression of NCALD improves proprioceptive contacts onto motoneuron somas [299]. A dual approach to treating SMA has been evaluated by combining SMN-increasing ASOs and NCALD-reducing ASOs in severe SMA model mice. Compound muscle action potentials, motor unit numbers, muscle fiber size, and grip strength were preserved when these two treatments were combined, compared to the use of SMN-ASOs alone [301]. Whether the endocytosis alterations (and thus ASO uptake into cells) resultant from NCALD downregulation contributes to the phenotypic improvement is unclear, however. In contrast to these positive modifying results, one investigation observed no association of NCALD and phenotypic modification unless multiple mutations of NCALD coincided to possibly create a cryptic splice site [294]. Thus, it is possible that NCALD-regulated modification is not widely applicable to most SMA-affected individuals with discordant phenotypes to SMN2 copy numbers.
PLS3 was the first reported positive modifier of the SMA phenotype, and was found to be highly upregulated in differentiated motoneurons obtained from fibroblasts of discordant siblings [290]. This report found that high levels of PLS3 protected individuals with 3–4 copies of SMN2 from SMA onset even in the presence of biallelic SMN1 deletion [290]. However, other studies debate the applicability of PLS3 protection across a large patient population. One recent publication utilized next-generation sequencing to evaluate PLS3 variants and found no correlation of PLS3 variants and phenotype, suggesting that PLS3-driven phenotypic modification may only occur in a small population of patients [297].
PLS3 is located on chromosome Xq23 and is a calcium-dependent F-actin-bundling protein that modulates the cytoskeleton, axonal growth and migration, vesicle trafficking, endocytosis, and regulates the ratio of G-actin to F-actin [302,303,304,305]. One study observed that overexpression of PLS3 improved the survival of mild and severe SMA model mice [298,302,306], while overexpression in SMN-deficient motoneurons and SMA morpholino zebrafish restored axonal growth and motor function [290,302,307]. It is noteworthy to repeat, however, that axon growth defects are not observed in SMA model mice [146,147] and thus putative modifiers likely do not alter axonal growth in humans. At the level of the spinal cord, PLS3 overexpression increased motoneuron soma size and the number of proprioceptive synapses in SMA model mice [80]. At the NMJ, PLS3 upregulation corresponded with augmented neurotransmission [80], restored endplate and muscle fiber size, improved vesicle trafficking and nerve terminal accumulation, restored endocytosis and actin dynamics, and increased the number of terminal active zones of SMA model mice and zebrafish [80,298,307]. Additionally, PLS3 was found to stabilize synaptic innervation, resulting in delayed axonal pruning in NMJs of SMA mice [80], thus improving the weakened nerve–muscle connection characteristic of SMA. Additionally, a study performed by Alrafiah et al. (2018) also observed reduced axonal defects in PLS3-upregulated motoneuron cultures, as well as improvements in lifespan of SMNΔ7 mice after PLS3 upregulation, although they did not observe sustained improvement in weight gain [302]. In contrast to these variable but positive benefits, however, an investigation by McGovern et al. (2015) observed no benefit to neuromuscular function, weight, lifespan or phenotype after PLS3 upregulation in SMNΔ7 mice [308]; Bowerman et al. (2009) also did not observe phenotypic improvement associated with elevated PLS3 levels [81]. Interestingly, Kaifer et al. (2017) observed benefits of PLS3 upregulation in mild SMA mouse models but not in severe SMNΔ7 mice [306]. The debate regarding PLS3 as a protective modifier remains unresolved, and further studies are required to understand which mechanism(s) or pathway(s) associated with PLS3 may modify the SMA phenotype.
PLS3 has several binding partners, one of which is the calcineurin inhibitor CHP1. CHP1 dephosphorylates proteins involved with calcineurin phosphatase activity and has elevated expression in SMA model mice. Knockdown of CHP1 restored axonal growth in Smn-depleted NCS34 motoneuron-like cells, SMA model zebrafish, and primary SMA model mouse motoneuron cultures [300]. In SMA model mice treated with SMN-based ASOs, CHP1 reduction prolonged survival, improved electrophysiological defects, NMJ growth and maturation, and muscle fiber size in comparison to the effects of ASOs alone. In addition to CHP1, PLS3 also binds to coronin-1C (a protein encoded by the CORO1C gene) in a calcium-dependent manner to mediate endocytosis and actin dynamics [298]. This evidence suggests that actin dynamics in motoneurons is a calcium-dependent process that strongly modulates disease pathogenesis by increasing neuromuscular function and stabilizing motoneuron circuitry.
9.5. Physical Therapy Strategies
The benefits of exercise have been evaluated in SMA model mice and SMA-affected individuals. In intermediate SMA model mice, elevated levels of full-length Smn have been observed after either acute or chronic exercise [309,310]. Chronic exercise significantly improved motoneuron maturation and soma loss in the lumbar spinal cord [311] and extended lifespan [310], suggesting that exercise can mitigate pathology and disease progression. Elevated levels of IGF-1 have been observed after exercise in SMA mice, potentially providing neuroprotective support through a trophic action rather than through muscle size improvement [312]. However, these trophic and SMN upregulation benefits were not observed in Type II patients performing arm cycling exercises [313]. Other studies evaluating the benefits of physical exercise in mild SMA model mice found improvements in glucose homeostasis, oxygen consumption, and muscle mitochondria function [314], especially when the exercise was high intensity. Chronic exercise in these mice improved muscle fatigue, neuromuscular excitability, and increased the resistance of muscles to damage [315].
Evaluation of exercise in patients has been primarily reported on Type II–III SMA patients not receiving SMN-based therapies. Rehabilitative interventions for SMA-affected individuals include physical therapy, strengthening and balance exercises, aquatic therapy, and physical activity. Most published reports on the benefits of exercise have been individual case studies [316], although some clinical trials have been initiated. For example, cycle ergometer training in Type III SMA patients has been demonstrated to improve oxidative capacity but induces significant fatigue [317]. Other programs have utilized at-home strength and aerobic exercise trainings to improve motor function, strength, fatigue, and cardiovascular fitness in patients. Benefits of these exercises (in particular, aerobic exercise) have been difficult to assess due to a high drop-out rate [318]. However, exercise through sport activity has been shown to significantly improve self-esteem and identity, reduce depression, and result in a greater quality of life for patients with neuromuscular disorders, including SMA-affected individuals [319]. With new FDA-approved SMN-based therapies improving the ability of SMA-affected individuals to participate in activity that demands endurance, strength, and motor skills, exercise may be an excellent, low-cost and accessible method to improve the negative psychological and emotional aspects of SMA.
9.6. Biomedical Devices
Advances in biomedical devices have shown that some SMA-affected individuals may benefit from use of orthoses (externally applied devices designed to improve muscular function), such as robotic exoskeletons. These devices improve quality of life for people with muscular diseases [320,321,322,323,324], including SMA. One study demonstrated positive effects on range of motion and performance of daily living activities of SMA-affected individuals [322]. Orthotic-assisted enhancement of motor function correlated with higher self-esteem, increased participation in school, and more social interaction [322], which result in a better quality of life. Another biomedical device, the robotic stretcher, leverages the retained function of limited digit movements to improve the operation of a motorized wheelchair, thus bettering independent maneuvering [325]. These devices have the ability to improve motor function for individuals who either do not experience significant improvement of motor function through other therapeutic means or are unable to utilize other treatments due to established weakness or paralysis, or due to lack of access. It will be interesting to see how these devices might further benefit the SMA population when used in conjunction with SMN-based genetic therapies.
10. Future Directions for SMA Therapies
The long-term success of SMA therapy depends on the extent of improvement and permanency of recovery. For most individuals, the best therapy to salvage motor function and lifespan will be SMN dependent. This conclusion is supported by compelling and vast evidence from animal models and patients demonstrating that SMN is crucial for motoneurons, and that restoration of SMN at any time point can provide (albeit sometimes limited) benefits to motor function. Current FDA-approved therapies for SMA (nusinersen, onasemnogene-xioi, and risdiplam) do not fully rescue motor impairment or development for all patients, particularly those who have only one copy of SMN2 or receive postsymptomatic treatment. Additionally, patients who are either ineligible for, or do not receive, SMN-based treatment (due to cost, availability, access, or condition) will require SMN-independent strategies to improve quality of life through better motor function.
Muscle strength and endurance (and consequently motor skill) are driven by neuromuscular activity. Motor skills permit the performance of activities required for daily living, including wheelchair mobility, daily tasks such as food preparation or hygienic practices, and the use of a keyboard and mouse. The ability to perform these activities would provide meaningful clinical improvement to patients and their caregivers [35,38]. SMN-based therapy is a remarkable start to improving motor function, but patients that respond suboptimally to treatment would benefit from complementary, SMN-independent medicine to further improve motor skills.
Finding a Cure through Complementary Treatment: NMJs Are Crucial Targets
Numerous reports demonstrate that NMJ instability is a crucial component of SMA pathogenesis [85,122,135,143,145,147,253,257,326,327], and evidence suggests that neuromuscular weakness persists after SMN-based treatment [46,279,280,327,328,329,330]. The potential discovery of SMA phenotypic modifiers further support the notion that the protection of NMJs can significantly modify disease progression [290,299,300]. Additionally, embryonic intervention to protect developing NMJs can improve the postnatal phenotype [253], suggesting that perinatal development is a critical window for optimal therapeutic benefit.
There remains a gap in effective SMA therapies that directly protect neuromuscular function despite the potential to drastically improve patient fatigue, independence, and quality of life [38]. For patients who receive suboptimal motor benefits from SMN-based medicines, protecting and improving neuromuscular function will be critical. One of the populations most vulnerable to persistent NMJ dysfunction after SMN-dependent treatments are adult SMA patients, as it is likely that some motoneuron loss has already occurred by the time SMN-based treatment is initiated.
One recent study that supports the need for NMJ-targeted treatment evaluated neuromuscular function after nusinersen treatment of adult SMA patients. Arnold and colleagues utilized repetitive nerve stimulation (3 Hz) and measured compound muscle action potential (CMAP) decrement to determine neuromuscular function. The investigators found that a large proportion of ambulatory and non-ambulatory adult SMA patients had improved CMAP amplitudes after 10–14 months of nusinersen treatment, but maintained a CMAP decrement (>10% at 3 Hz) upon repetitive nerve stimulation [330], indicating that neuromuscular transmission defects persist after nusinersen treatment. Additionally, the investigators observed a correlation between CMAP decrement and motor function (measured using 6 min walk test and fatigue, elbow extension/flexion, shoulder abduction and revised upper limb module). These data suggest that neuromuscular defects in adult SMA patients constitute a secondary pathology that is not improved by SMN-based strategies. One possible explanation for this observation is that neuromuscular denervation persists after SMN restoration but can be compensated for by collateral sprouting. Additionally, the investigators did not observe a correlation between CMAP decrement and disease severity (ambulatory or non-ambulatory function), duration of disease (years since symptom onset), or patient age [330]. Thus, individual differences in NMJ transmission may be an SMN-independent modifier of disease phenotype [330]. Understanding the mechanisms underlying neuromuscular dysfunction will be crucial for restoring motor ability and strength in patients receiving SMN-based therapy.
Similar results investigating neuromuscular pathology before and after ISS-N1 ASO treatment have also been observed in SMNΔ7 mice. One study noted a similar persistence of CMAP decrement (measured during adulthood) after P4 ASO treatment in SMNΔ7 mice [330]. Another study demonstrated that P0-1 ASO treatment of SMNΔ7 mice did not restore neuromuscular transmission in a highly vulnerable muscle (transverse abdominis). Furthermore, the investigators also observed that increased presynaptic transmitter release (via drug-induced augmented calcium influx into motor nerve terminals) in vivo resulted in greater in vivo muscle strength in SMNΔ7 mice [279,280], further supporting the notion that neuromuscular transmission correlates with motor function in SMA. Together, these studies indicate that established neuromuscular pathology is unlikely to be reversed by SMN-based medicine and thus complementary therapies are needed to optimally stabilize and strengthen neuromuscular connections and thus maximize motor ability. This goal might be achieved by use of drugs that increase presynaptic neurotransmitter release, alter postsynaptic excitability, and/or enhance response to neurotransmitter, which can increase neurotrophic support [331] as well as increase the number of NMJs firing for a particular stimulus rate and consequently induce stronger muscle contractions.
As individuals receiving SMN-based therapy(s) progress past natural disorder outcomes, new insight will potentially reveal previously obfuscated or residual pathologies that require targeted treatments to improve quality of life. In order to develop a therapeutic cure for SMA, complementary SMN-dependent and -independent treatment strategies are necessary to address all aspects of SMA pathology to improve quality of life across the lifespan of SMA-affected individuals.
Author Contributions
Conceptualization, K.S.O., E.J.R., C.J.D., and S.D.M.; writing—original draft preparation, K.S.O. and E.J.R.; writing—review and editing, K.S.O., E.J.R., C.J.D., and S.D.M.; visualization, K.S.O., E.J.R., C.J.D., and S.D.M.; funding acquisition, K.S.O., C.J.D., and S.D.M. All authors have read and agreed to the published version of the manuscript.
Funding
K.S.O. is supported by funding from NIH NINDS F31NS196753. C.J.D. is supported by funding from NIH NINDS R01NS060926, R21NS103107, the Muscular Dystrophy Association (MDA418685) and CureSMA (DID1617 and 1718). S.D.M. is supported by grants from the Muscular Dystrophy Associate (MDA603852) and CureSMA (MER2021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.; Androphy, E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997, 61, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef] [Green Version]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, B.T. Spinal muscular atrophies. Pediatr. Clin. N. Am. 2015, 62, 743–766. [Google Scholar] [CrossRef]
- Faravelli, I.; Nizzardo, M.; Comi, G.P.; Corti, S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat. Rev. Neurol. 2015, 11, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Munsat, T.L. Workshop Report: International SMA Collaboration. Neuromuscul. Disord. 1991, 1, 81. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef]
- Zerres, K.; Davies, K.E. 59th ENMC International Workshop: Spinal Muscular Atrophies: Recent progress and revised diagnostic criteria, 17–19 April 1998, Soestduinen, The Netherlands. Neuromuscul. Disord. 1999, 9, 272–278. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef]
- Darras, B.T.; Finkel, R.S. Natural History of Spinal Muscular Atrophy. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 399–421. [Google Scholar]
- Dubowitz, V. Very severe spinal muscular atrophy (SMA type 0): An expanding clinical phenotype. Eur. J. Paediatr. Neurol. 1999, 3, 49–51. [Google Scholar] [CrossRef]
- MacLeod, M.J.; Taylor, J.E.; Lunt, P.W.; Mathew, C.G.; Robb, S.A. Prenatal onset spinal muscular atrophy. Eur. J. Paediatr. Neurol. 1999, 3, 65–72. [Google Scholar] [CrossRef]
- Rudnik-Schoneborn, S.; Vogelgesang, S.; Armbrust, S.; Graul-Neumann, L.; Fusch, C.; Zerres, K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve 2010, 42, 144–147. [Google Scholar] [CrossRef]
- Carrasco, D.; Magoulas, P.; Scull, J.C.; Jarrell, J.A.; Lalani, S.R.; Wangler, M.F. Digital necrosis in an infant with severe spinal muscular atrophy. Neurol. Genet. 2019, 5, e361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnik-Schoneborn, S.; Goebel, H.H.; Schlote, W.; Molaian, S.; Omran, H.; Ketelsen, U.; Korinthenberg, R.; Wenzel, D.; Lauffer, H.; Kreiss-Nachtsheim, M.; et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology 2003, 60, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schoneborn, S.; Heller, R.; Berg, C.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Zerres, K. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. 2008, 45, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Harding, B.N.; Kariya, S.; Monani, U.R.; Chung, W.K.; Benton, M.; Yum, S.W.; Tennekoon, G.; Finkel, R.S. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J. Neuropathol. Exp. Neurol. 2015, 74, 15–24. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schoneborn, S.; Forrest, E.; Lusakowska, A.; Borkowska, J.; Hausmanowa-Petrusewicz, I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J. Neurol. Sci. 1997, 146, 67–72. [Google Scholar] [CrossRef]
- Montes, J.; McDermott, M.P.; Mirek, E.; Mazzone, E.S.; Main, M.; Glanzman, A.M.; Duong, T.; Young, S.D.; Salazar, R.; Pasternak, A.; et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression. PLoS ONE 2018, 13, e0199657. [Google Scholar] [CrossRef]
- Piepers, S.; van den Berg, L.H.; Brugman, F.; Scheffer, H.; Ruiterkamp-Versteeg, M.; van Engelen, B.G.; Faber, C.G.; de Visser, M.; van der Pol, W.L.; Wokke, J.H. A natural history study of late onset spinal muscular atrophy types 3b and 4. J. Neurol. 2008, 255, 1400–1404. [Google Scholar] [CrossRef]
- Mongiovi, P.; Dilek, N.; Garland, C.; Hunter, M.; Kissel, J.T.; Luebbe, E.; McDermott, M.P.; Johnson, N.; Heatwole, C. Patient Reported Impact of Symptoms in Spinal Muscular Atrophy (PRISM-SMA). Neurology 2018, 91, e1206–e1214. [Google Scholar] [CrossRef]
- Wan, H.W.Y.; Carey, K.A.; D’Silva, A.; Vucic, S.; Kiernan, M.C.; Kasparian, N.A.; Farrar, M.A. Health, wellbeing and lived experiences of adults with SMA: A scoping systematic review. Orphanet J. Rare Dis. 2020, 15, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Bastida, J.; Pena-Longobardo, L.M.; Aranda-Reneo, I.; Tizzano, E.; Sefton, M.; Oliva-Moreno, J. Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J. Rare Dis. 2017, 12, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjorth, E.; Kreicbergs, U.; Sejersen, T.; Lovgren, M. Parents’ advice to healthcare professionals working with children who have spinal muscular atrophy. Eur. J. Paediatr. Neurol. 2018, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; McGraw, S.; Henne, J.; Jarecki, J.; Hobby, K.; Yeh, W.S. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: A qualitative study. BMC Neurol. 2015, 15, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, R.; Belter, L.; Wasnock, M.; Nazarelli, A.; Jarecki, J. Evaluating Benefit-risk Decision-making in Spinal Muscular Atrophy: A First-ever Study to Assess Risk Tolerance in the SMA Patient Community. Clin. Ther. 2019, 41, 943–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGraw, S.; Qian, Y.; Henne, J.; Jarecki, J.; Hobby, K.; Yeh, W.S. A qualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. 2017, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Lucibello, S.; Perulli, M.; Coratti, G.; de Sanctis, R.; Pera, M.C.; Pane, M.; Montes, J.; de Vivo, D.C.; Darras, B.T.; et al. Longitudinal natural history of type I spinal muscular atrophy: A critical review. Orphanet J. Rare Dis. 2020, 15, 84. [Google Scholar] [CrossRef]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child Neurol. 2007, 22, 1027–1049. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Nurputra, D.K.; Lai, P.S.; Harahap, N.I.; Morikawa, S.; Yamamoto, T.; Nishimura, N.; Kubo, Y.; Takeuchi, A.; Saito, T.; Takeshima, Y.; et al. Spinal muscular atrophy: From gene discovery to clinical trials. Ann. Hum. Genet. 2013, 77, 435–463. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Day, J.W.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.; Schultz, M.; et al. AVXS-101 phase 3 study in spinal muscular atrophy type 1. J. Neurol. Neurosurg. Psychiatry 2019, 90, e8. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 2020, 1, CD006282. [Google Scholar] [CrossRef] [PubMed]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [Green Version]
- Luhrmann, R.; Kastner, B.; Bach, M. Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim. Biophys. Acta 1990, 1087, 265–292. [Google Scholar] [CrossRef]
- Lerner, M.R.; Steitz, J.A. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1979, 76, 5495–5499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatnik, A.J.; McGovern, V.L.; Le, T.T.; Iyer, C.C.; Kaspar, B.K.; Burghes, A.H.M. Conditional deletion of SMN in cell culture identifies functional SMN alleles. Hum. Mol. Genet. 2021, 29, 3477–3492. [Google Scholar] [CrossRef]
- So, B.R.; Zhang, Z.; Dreyfuss, G. The Function of Survival Motor Neuron Complex and Its Role in Spinal Muscular Atrophy Pathogenesis. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 99–111. [Google Scholar]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Pellizzoni, L.; Kataoka, N.; Charroux, B.; Dreyfuss, G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 1998, 95, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Baccon, J.; Pellizzoni, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J. Biol. Chem. 2002, 277, 31957–31962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhler, D.; Raker, V.; Luhrmann, R.; Fischer, U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: Implications for spinal muscular atrophy. Hum. Mol. Genet. 1999, 8, 2351–2357. [Google Scholar] [CrossRef]
- Carissimi, C.; Baccon, J.; Straccia, M.; Chiarella, P.; Maiolica, A.; Sawyer, A.; Rappsilber, J.; Pellizzoni, L. Unrip is a component of SMN complexes active in snRNP assembly. FEBS Lett. 2005, 579, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Carissimi, C.; Saieva, L.; Baccon, J.; Chiarella, P.; Maiolica, A.; Sawyer, A.; Rappsilber, J.; Pellizzoni, L. Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J. Biol. Chem. 2006, 281, 8126–8134. [Google Scholar] [CrossRef] [Green Version]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J. Cell Biol. 1999, 147, 1181–1194. [Google Scholar] [CrossRef]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Yong, J.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin4: A novel component of the SMN complex that is found in both gems and nucleoli. J. Cell Biol. 2000, 148, 1177–1186. [Google Scholar] [CrossRef]
- Fischer, U.; Liu, Q.; Dreyfuss, G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997, 90, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Gubitz, A.K.; Mourelatos, Z.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J. Biol. Chem. 2002, 277, 5631–5636. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Buhler, D.; Pillai, R.; Lottspeich, F.; Fischer, U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol 2001, 3, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Baccon, J.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J. Biol. Chem. 2002, 277, 7540–7545. [Google Scholar] [CrossRef] [Green Version]
- Pellizzoni, L.; Yong, J.; Dreyfuss, G. Essential role for the SMN complex in the specificity of snRNP assembly. Science 2002, 298, 1775–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. Spinal muscular atrophy: The role of SMN in axonal mRNA regulation. Brain Res. 2012, 1462, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; Vercelli, A. Neuromuscular Junctions as Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef] [Green Version]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Goncalves Ido, C.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Rathod, R.; Havlicek, S.; Frank, N.; Blum, R.; Sendtner, M. Laminin induced local axonal translation of beta-actin mRNA is impaired in SMN-deficient motoneurons. Histochem. Cell Biol. 2012, 138, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.M.; Shen, H.; Barry, D.M.; Garcia, V.B.; Rose, F.F., Jr.; Lorson, C.L.; Garcia, M.L. The spinal muscular atrophy mouse model, SMADelta7, displays altered axonal transport without global neurofilament alterations. Acta Neuropathol. 2011, 122, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Sasaki, Y.; Wen, Z.; Bassell, G.J.; Zheng, J.Q. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat. Neurosci. 2006, 9, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, B.; Krober, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Hosseini Barkooie, S.M.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013, 22, 1328–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowerman, M.; Anderson, C.L.; Beauvais, A.; Boyl, P.P.; Witke, W.; Kothary, R. SMN, profilin IIa and plastin 3: A link between the deregulation of actin dynamics and SMA pathogenesis. Mol. Cell. Neurosci. 2009, 42, 66–74. [Google Scholar] [CrossRef]
- Giesemann, T.; Rathke-Hartlieb, S.; Rothkegel, M.; Bartsch, J.W.; Buchmeier, S.; Jockusch, B.M.; Jockusch, H. A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J. Biol. Chem. 1999, 274, 37908–37914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bergeijk, J.; Rydel-Konecke, K.; Grothe, C.; Claus, P. The spinal muscular atrophy gene product regulates neurite outgrowth: Importance of the C terminus. FASEB J. 2007, 21, 1492–1502. [Google Scholar] [CrossRef]
- Fan, L.; Simard, L.R. Survival motor neuron (SMN) protein: Role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 2002, 11, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef]
- Gogliotti, R.G.; Quinlan, K.A.; Barlow, C.B.; Heier, C.R.; Heckman, C.J.; Didonato, C.J. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 2012, 32, 3818–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, T.L.; Kong, L.; Wang, X.; Osborne, M.A.; Crowder, M.E.; Van Meerbeke, J.P.; Xu, X.; Davis, C.; Wooley, J.; Goldhamer, D.J.; et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 2012, 32, 8703–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, V.L.; Iyer, C.C.; Arnold, W.D.; Gombash, S.E.; Zaworski, P.G.; Blatnik, A.J., 3rd; Foust, K.D.; Burghes, A.H. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNDelta7 mouse model of SMA. Hum. Mol. Genet. 2015, 24, 5524–5541. [Google Scholar] [CrossRef] [Green Version]
- Paez-Colasante, X.; Seaberg, B.; Martinez, T.L.; Kong, L.; Sumner, C.J.; Rimer, M. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS ONE 2013, 8, e75866. [Google Scholar] [CrossRef] [Green Version]
- Park, G.H.; Maeno-Hikichi, Y.; Awano, T.; Landmesser, L.T.; Monani, U.R. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J. Neurosci. 2010, 30, 12005–12019. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Coovert, D.D.; Burghes, A.H. Animal models of spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 2451–2457. [Google Scholar] [CrossRef] [Green Version]
- Gabanella, F.; Butchbach, M.E.; Saieva, L.; Carissimi, C.; Burghes, A.H.; Pellizzoni, L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabanella, F.; Carissimi, C.; Usiello, A.; Pellizzoni, L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 2005, 14, 3629–3642. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.M.; Kariya, S.; Patruni, S.; Osborne, M.A.; Liu, D.; Henderson, C.E.; Li, D.K.; Pellizzoni, L.; Rojas, J.; Valenzuela, D.M.; et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Investig. 2011, 121, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Burlet, P.; Huber, C.; Bertrandy, S.; Ludosky, M.A.; Zwaenepoel, I.; Clermont, O.; Roume, J.; Delezoide, A.L.; Cartaud, J.; Munnich, A.; et al. The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum. Mol. Genet. 1998, 7, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Schrank, B.; Kralewski, M.; Rossoll, W.; Sendtner, M. Reduced survival motor neuron (Smn) gene dose in mice leads to motor neuron degeneration: An animal model for spinal muscular atrophy type III. Hum. Mol. Genet. 2000, 9, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.K.; Gibbs, R.M.; Feng, Z.; Ko, C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012, 21, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Sendtner, M. Developmental regulation of SMN expression: Pathophysiological implications and perspectives for therapy development in spinal muscular atrophy. Gene Ther. 2017, 24, 506–513. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; DiDonato, C.J.; McGovern, V.L.; Arnold, W.D. Mammalian Models of Spinal Muscular Atrophy. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 241–260. [Google Scholar]
- Gavrilina, T.O.; McGovern, V.L.; Workman, E.; Crawford, T.O.; Gogliotti, R.G.; DiDonato, C.J.; Monani, U.R.; Morris, G.E.; Burghes, A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008, 17, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Fu, A.K.; Ip, N.Y. Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 2012, 35, 441–453. [Google Scholar] [CrossRef]
- Duque, S.I.; Arnold, W.D.; Odermatt, P.; Li, X.; Porensky, P.N.; Schmelzer, L.; Meyer, K.; Kolb, S.J.; Schumperli, D.; Kaspar, B.K.; et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 2015, 77, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Murray, L.M.; Beauvais, A.; Pinheiro, B.; Kothary, R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 2012, 22, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Gogliotti, R.G.; Cardona, H.; Singh, J.; Bail, S.; Emery, C.; Kuntz, N.; Jorgensen, M.; Durens, M.; Xia, B.; Barlow, C.; et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013, 22, 4084–4101. [Google Scholar] [CrossRef] [Green Version]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; DiDonato, C.J. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Michaud, M.; Arnoux, T.; Bielli, S.; Durand, E.; Rotrou, Y.; Jablonka, S.; Robert, F.; Giraudon-Paoli, M.; Riessland, M.; Mattei, M.G.; et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 2010, 38, 125–135. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.M.; Beauvais, A.; Gibeault, S.; Courtney, N.L.; Kothary, R. Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun. 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [Green Version]
- Nichterwitz, S.; Nijssen, J.; Storvall, H.; Schweingruber, C.; Comley, L.H.; Allodi, I.; Lee, M.V.; Deng, Q.; Sandberg, R.; Hedlund, E. LCM-seq reveals unique transcriptional adaptation mechanisms of resistant neurons and identifies protective pathways in spinal muscular atrophy. Genome Res. 2020, 30, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Broner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef]
- Fayzullina, S.; Martin, L.J. Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of Spinal Muscular Atrophy (SMA). PLoS ONE 2014, 9, e93329. [Google Scholar] [CrossRef] [PubMed]
- Huo, Q.; Kayikci, M.; Odermatt, P.; Meyer, K.; Michels, O.; Saxena, S.; Ule, J.; Schumperli, D. Splicing changes in SMA mouse motoneurons and SMN-depleted neuroblastoma cells: Evidence for involvement of splicing regulatory proteins. RNA Biol. 2014, 11, 1430–1446. [Google Scholar] [CrossRef] [Green Version]
- Baumer, D.; Lee, S.; Nicholson, G.; Davies, J.L.; Parkinson, N.J.; Murray, L.M.; Gillingwater, T.H.; Ansorge, O.; Davies, K.E.; Talbot, K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009, 5, e1000773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O’Donovan, M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, K.A.; Reedich, E.J.; Arnold, W.D.; Puritz, A.C.; Cavarsan, C.F.; Heckman, C.J.; DiDonato, C.J. Hyperexcitability precedes motoneuron loss in the Smn(2B/-) mouse model of spinal muscular atrophy. J. Neurophysiol. 2019, 122, 1297–1311. [Google Scholar] [CrossRef]
- Ling, K.K.; Lin, M.Y.; Zingg, B.; Feng, Z.; Ko, C.P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE 2010, 5, e15457. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.; Pearson, K.G. The role of proprioceptive feedback in the regulation and adaptation of locomotor activity. Adv. Exp. Med. Biol. 2002, 508, 343–355. [Google Scholar] [CrossRef]
- Windhorst, U. Muscle proprioceptive feedback and spinal networks. Brain Res. Bull. 2007, 73, 155–202. [Google Scholar] [CrossRef] [PubMed]
- Davidoff, R.A. Skeletal muscle tone and the misunderstood stretch reflex. Neurology 1992, 42, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Dietz, V.; Sinkjaer, T. Spastic movement disorder: Impaired reflex function and altered muscle mechanics. Lancet Neurol. 2007, 6, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Bhatia, K.; Kannan, A.; Gangwani, L. Molecular Mechanisms of Neurodegeneration in Spinal Muscular Atrophy. J. Exp. Neurosci. 2016, 10, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 6986–7004. [Google Scholar] [CrossRef] [Green Version]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of p53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576. [Google Scholar] [CrossRef] [Green Version]
- Courtney, N.L.; Mole, A.J.; Thomson, A.K.; Murray, L.M. Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death Dis. 2019, 10, 515. [Google Scholar] [CrossRef]
- Reedich, E.J.; Kalski, M.; Armijo, N.; Cox, G.A.; DiDonato, C.J. Spinal motor neuron loss occurs through a p53-and-p21-independent mechanism in the Smn2B/− mouse model of spinal muscular atrophy. Exp. Neurol. 2021, 337, 113587. [Google Scholar] [CrossRef]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Park, G.H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marce, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef]
- Neve, A.; Trub, J.; Saxena, S.; Schumperli, D. Central and peripheral defects in motor units of the diaphragm of spinal muscular atrophy mice. Mol. Cell. Neurosci. 2016, 70, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Torres-Benito, L.; Neher, M.F.; Cano, R.; Ruiz, R.; Tabares, L. SMN requirement for synaptic vesicle, active zone and microtubule postnatal organization in motor nerve terminals. PLoS ONE 2011, 6, e26164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, R.; Lopez-Manzaneda, M.; Arumugam, S.; Tabares, L. Synaptotagmin-2, and -1, linked to neurotransmission impairment and vulnerability in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 4703–4716. [Google Scholar] [CrossRef] [PubMed]
- Voigt, T.; Meyer, K.; Baum, O.; Schumperli, D. Ultrastructural changes in diaphragm neuromuscular junctions in a severe mouse model for Spinal Muscular Atrophy and their prevention by bifunctional U7 snRNA correcting SMN2 splicing. Neuromuscul. Disord. 2010, 20, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.C.; Denton, K.R.; Wang, Z.B.; Zhang, X.; Li, X.J. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis. Model. Mech. 2016, 9, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.G.; Deguise, M.O.; Murray, L.M.; Yazdani, A.; De Repentigny, Y.; Boudreau-Lariviere, C.; Kothary, R. Myogenic program dysregulation is contributory to disease pathogenesis in spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 4249–4259. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R.; Casanas, J.J.; Torres-Benito, L.; Cano, R.; Tabares, L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010, 30, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, R.; Tabares, L. Neurotransmitter release in motor nerve terminals of a mouse model of mild spinal muscular atrophy. J. Anat. 2014, 224, 74–84. [Google Scholar] [CrossRef]
- Jablonka, S.; Beck, M.; Lechner, B.D.; Mayer, C.; Sendtner, M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007, 179, 139–149. [Google Scholar] [CrossRef] [Green Version]
- McGovern, V.L.; Gavrilina, T.O.; Beattie, C.E.; Burghes, A.H. Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet. 2008, 17, 2900–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.L.; Chen, T.H.; Hsu, Y.Y.; Cheng, Y.H.; Juang, B.T.; Jong, Y.J. Selective Neuromuscular Denervation in Taiwanese Severe SMA Mouse Can Be Reversed by Morpholino Antisense Oligonucleotides. PLoS ONE 2016, 11, e0154723. [Google Scholar] [CrossRef]
- Thomson, S.R.; Nahon, J.E.; Mutsaers, C.A.; Thomson, D.; Hamilton, G.; Parson, S.H.; Gillingwater, T.H. Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLoS ONE 2012, 7, e52605. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Croizat, B.; Lagrange, M.C.; Warter, J.M.; Poindron, P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet 1995, 345, 694–695. [Google Scholar] [CrossRef]
- Walker, M.P.; Rajendra, T.K.; Saieva, L.; Fuentes, J.L.; Pellizzoni, L.; Matera, A.G. SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet. 2008, 17, 3399–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berciano, M.T.; Castillo-Iglesias, M.S.; Val-Bernal, J.F.; Lafarga, V.; Rodriguez-Rey, J.C.; Lafarga, M.; Tapia, O. Mislocalization of SMN from the I-band and M-band in human skeletal myofibers in spinal muscular atrophy associates with primary structural alterations of the sarcomere. Cell Tissue Res. 2020, 381, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.P.; Monani, U.R. Muscle-specific SMN reduction reveals motor neuron-independent disease in spinal muscular atrophy models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef] [Green Version]
- Iyer, C.C.; McGovern, V.L.; Murray, J.D.; Gombash, S.E.; Zaworski, P.G.; Foust, K.D.; Janssen, P.M.; Burghes, A.H. Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNDelta7 mouse model of SMA. Hum. Mol. Genet. 2015, 24, 6160–6173. [Google Scholar] [CrossRef] [Green Version]
- Guettier-Sigrist, S.; Coupin, G.; Braun, S.; Rogovitz, D.; Courdier, I.; Warter, J.M.; Poindron, P. On the possible role of muscle in the pathogenesis of spinal muscular atrophy. Fundam. Clin. Pharmacol. 2001, 15, 31–40. [Google Scholar] [CrossRef]
- Shafey, D.; Cote, P.D.; Kothary, R. Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 2005, 311, 49–61. [Google Scholar] [CrossRef]
- Arnold, A.S.; Gueye, M.; Guettier-Sigrist, S.; Courdier-Fruh, I.; Coupin, G.; Poindron, P.; Gies, J.P. Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab. Investig. 2004, 84, 1271–1278. [Google Scholar] [CrossRef]
- Bricceno, K.V.; Martinez, T.; Leikina, E.; Duguez, S.; Partridge, T.A.; Chernomordik, L.V.; Fischbeck, K.H.; Sumner, C.J.; Burnett, B.G. Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Hum. Mol. Genet. 2014, 23, 4745–4757. [Google Scholar] [CrossRef] [Green Version]
- Hayhurst, M.; Wagner, A.K.; Cerletti, M.; Wagers, A.J.; Rubin, L.L. A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Dev. Biol. 2012, 368, 323–334. [Google Scholar] [CrossRef]
- Osborne, M.; Gomez, D.; Feng, Z.; McEwen, C.; Beltran, J.; Cirillo, K.; El-Khodor, B.; Lin, M.Y.; Li, Y.; Knowlton, W.M.; et al. Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum. Mol. Genet. 2012, 21, 4431–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.I.; Mikesh, M.; Smith, I.; Rimer, M.; Thompson, W. Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev. Biol. 2011, 356, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.; Aghamaleky Sarvestany, A.; Roche, S.L.; Symes, R.C.; Gillingwater, T.H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 2235–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, G.; Powis, R.A.; Jones, R.A.; Groen, E.J.; Shorrock, H.K.; Lane, F.M.; Zheng, Y.; Sherman, D.L.; Brophy, P.J.; Gillingwater, T.H. Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 2853–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindt, H.; Feng, Z.; Mazzasette, C.; Glascock, J.J.; Valdivia, D.; Pyles, N.; Crawford, T.O.; Swoboda, K.J.; Patitucci, T.N.; Ebert, A.D.; et al. Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 4094–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.A.; Stucky, C.L.; Ebert, A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Araujo, A.; Araujo, M.; Swoboda, K.J. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J. Pediatr. 2009, 155, 292–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, J.; Martinez-Hernandez, R.; Also-Rallo, E.; Alias, L.; Barcelo, M.J.; Amenedo, M.; Medina, C.; Senosiain, R.; Calaf, J.; Baiget, M.; et al. Ultrasound evaluation of fetal movements in pregnancies at risk for severe spinal muscular atrophy. Neuromuscul. Disord. 2011, 21, 97–101. [Google Scholar] [CrossRef]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Biondi, O.; Lopes, P.; Desseille, C.; Branchu, J.; Chali, F.; Ben Salah, A.; Pariset, C.; Chanoine, C.; Charbonnier, F. Physical exercise reduces cardiac defects in type 2 spinal muscular atrophy-like mice. J. Physiol. 2012, 590, 5907–5925. [Google Scholar] [CrossRef]
- Bevan, A.K.; Hutchinson, K.R.; Foust, K.D.; Braun, L.; McGovern, V.L.; Schmelzer, L.; Ward, J.G.; Petruska, J.C.; Lucchesi, P.A.; Burghes, A.H.; et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010, 19, 3895–3905. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.R.; DiDonato, C.J. ECG in neonate mice with spinal muscular atrophy allows assessment of drug efficacy. Front. Biosci. (Elite Ed.) 2015, 7, 107–116. [Google Scholar]
- Heier, C.R.; Satta, R.; Lutz, C.; DiDonato, C.J. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 2010, 19, 3906–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Maxwell, G.K.; Szunyogova, E.; Shorrock, H.K.; Gillingwater, T.H.; Parson, S.H. Developmental and degenerative cardiac defects in the Taiwanese mouse model of severe spinal muscular atrophy. J. Anat. 2018, 232, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreml, J.; Riessland, M.; Paterno, M.; Garbes, L.; Rossbach, K.; Ackermann, B.; Kramer, J.; Somers, E.; Parson, S.H.; Heller, R.; et al. Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 2013, 21, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Habibi, J.; Ma, L.; Glascock, J.J.; Sowers, J.R.; Lorson, C.L. Partial restoration of cardio-vascular defects in a rescued severe model of spinal muscular atrophy. J. Mol. Cell. Cardiol. 2012, 52, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Habibi, J.; Yang, H.T.; Vale, S.M.; Sewell, W.A.; Lorson, C.L. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 2010, 19, 4059–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Wan, B.; Feng, P.; Sun, J.; Rigo, F.; Bennett, C.F.; Akerman, M.; Krainer, A.R.; Hua, Y. Downregulation of Survivin contributes to cell-cycle arrest during postnatal cardiac development in a severe spinal muscular atrophy mouse model. Hum. Mol. Genet. 2018, 27, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Chen, C.L.; Ting, C.H.; Lin-Chao, S.; Hwu, W.L.; Dodge, J.C.; Passini, M.A.; Cheng, S.H. Systemic administration of a recombinant AAV1 vector encoding IGF-1 improves disease manifestations in SMA mice. Mol. Ther. 2014, 22, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Kumada, S.; Uchiyama, A.; Saito, K.; Osawa, M.; Yagishita, A.; Kurata, K.; Hayashi, M. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings. Brain Dev. 2004, 26, 53–56. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Shababi, M.; Lorson, C.L.; Rudnik-Schoneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell. Mol. Life Sci. 2016, 73, 1003–1020. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.-P.; Wang, G.-S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as Just a Motor Neuron Disease. Pediatr. Neurol. 2020, 109, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [Green Version]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Chen, T.H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.S.; Jarecki, J.; MacKenzie, A.; Chen, K.S. Spinal Muscular Atrophy Therapeutics Development. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 263–281. [Google Scholar]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef]
- Singh, N.N.; Lee, B.M.; DiDonato, C.J.; Singh, R.N. Mechanistic principles of antisense targets for the treatment of spinal muscular atrophy. Future Med. Chem. 2015, 7, 1793–1808. [Google Scholar] [CrossRef] [Green Version]
- Verma, A. Recent Advances in Antisense Oligonucleotide Therapy in Genetic Neuromuscular Diseases. Ann. Indian Acad. Neurol. 2018, 21, 3–8. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O’Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef] [Green Version]
- D’Ydewalle, C.; Sumner, C.J. Spinal Muscular Atrophy Therapeutics: Where do we Stand? Neurotherapeutics 2015, 12, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Ramos, D.M.; d’Ydewalle, C.; Gabbeta, V.; Dakka, A.; Klein, S.K.; Norris, D.A.; Matson, J.; Taylor, S.J.; Zaworski, P.G.; Prior, T.W.; et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J. Clin. Investig. 2019, 129, 4817–4831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuluaga Sanchez, S.; Purser, M.; Mader, G.; Gould, I.G.; Knight, C.; Johnson, N.B.; Patel, M.; Jensen, J.; Odom, T. Improved quality of life and life-years in patients with infantile-onset SMA following treatment with nusinersen. Value Health 2019, 22, S337. [Google Scholar] [CrossRef]
- Hagenacker, T.; Wurster, C.D.; Gunther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef]
- Jochmann, E.; Steinbach, R.; Jochmann, T.; Chung, H.Y.; Rodiger, A.; Neumann, R.; Mayer, T.E.; Kirchhof, K.; Loudovici-Krug, D.; Smolenski, U.C.; et al. Experiences from treating seven adult 5q spinal muscular atrophy patients with Nusinersen. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420907803. [Google Scholar] [CrossRef] [Green Version]
- Arnold, W.; McGovern, V.L.; Sanchez, B.; Li, J.; Corlett, K.M.; Kolb, S.J.; Rutkove, S.B.; Burghes, A.H. The neuromuscular impact of symptomatic SMN restoration in a mouse model of spinal muscular atrophy. Neurobiol. Dis. 2016, 87, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Crawford, T.; Sumner, C.; Finkel, R.; De Vivo, D.; Oskoui, M.; Tizzano, E.; Zhao, G.; Petrillo, M.; Stebbins, C.; Farwell, W. Phosphorylated neurofilament heavy chain (pNF-H) levels in infants and children with SMA: Evaluation of pNF-H as a potential biomarker of SMA disease activity. Neuromuscul. Disord. 2018, 28, S110–S111. [Google Scholar] [CrossRef]
- Darras, B.; Finkel, R.; Mercuri, E.; Sumner, C.; Oskoui, M.; Tizzano, E.; Ryan, M.; Zhao, G.; Petrillo, M.; Stebbins, C.; et al. Association of phosphorylated neurofilament heavy chain (pNF-H) with nusinersen treatment of SMA: Analyses from the ENDEAR and CHERISH studies. Neuromuscul. Disord. 2018, 28, S31. [Google Scholar] [CrossRef]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porensky, P.N.; Burghes, A.H. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum. Gene Ther. 2013, 24, 489–498. [Google Scholar] [CrossRef] [Green Version]
- SPINRAZA [Package Insert]; Biogen Inc.: Cambridge, MA, USA, 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf (accessed on 3 February 2021).
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Schmelzer, L.; Braun, L.; McGovern, V.; Likhite, S.; Michels, O.; Govoni, A.; Fitzgerald, J.; Morales, P.; et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: A dose-response study in mice and nonhuman primates. Mol. Ther. 2015, 23, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Crawford, T.O. Two breakthrough gene-targeted treatments for spinal muscular atrophy: Challenges remain. J. Clin. Investig. 2018, 128, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- ZOLGENSMA [Package Insert]; AveXis, Inc.: Bannockburn, IL, USA, 2019. Available online: https://www.fda.gov/media/126109/download (accessed on 3 February 2021).
- Dyer, O. Health ministers condemn Novartis lottery for Zolgensma, the world’s most expensive drug. BMJ 2020, 368, m580. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; Servais, L.; Day, J.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 16-month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with type 1 spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef]
- Mercuri, E.; Barisic, N.; Boespflug-Tanguy, O.; Deconinck, N.; Kostera-Pruszczyk, A.; Masson, R.; Mazzone, E.; Nascimento, A.; Saito, K.; Vlodavets, D.; et al. SUNFISH Part 2: Efficacy and Safety of Risdiplam (RG7916) in Patients with Type 2 or Non-Ambulant Type 3 Spinal Muscular Atrophy (SMA) (1260). Neurology 2020, 94, 1260. [Google Scholar]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Genentech Inc. Evrysdi™ (Risdiplam): US Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213535s000lbl.pdf (accessed on 3 February 2021).
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Pagliarulo, N. Zolgensma Set a New Drug Pricing Bar. Insurers Show Some Signs of Pushback. BioPharma Dive. 2019. Available online: https://www.biopharmadive.com/news/zolgensma-set-a-new-drug-pricing-bar-insurers-show-some-signs-of-pushback/558101/ (accessed on 3 February 2021).
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Dixon, S.; Naik, R.; Noone, J.M.; Buchenberger, J.D.; Whitmire, S.M.; Mills, R.; Arnold, W. Early experiences of nusinersen for the treatment of spinal muscular atrophy: Results from a large survey of patients and caregivers. Muscle Nerve 2020, 1–9. [Google Scholar] [CrossRef]
- Ellis, A.G.; Mickle, K.; Herron-Smith, S.; Kumar, V.M.; Cianciolo, L.; Seidner, M.; Thokala, P.; Stevenson, M.; Rind, D.; Pearson, S.D. Spinraza® and Zolgensma® for Spinal Muscular Atrophy: Effectiveness and Value; Institute for Clinical and Economic Review: Boston, MA, USA, 2019. [Google Scholar]
- Starner, C.I.; Gleason, P.P. Spinal Muscular Atrophy Therapies: ICER Grounds the Price to Value Conversation in Facts. J. Manag. Care Spec. Pharm. 2019, 25, 1306–1308. [Google Scholar] [CrossRef]
- Darras, B.T.; De Vivo, D.C. Precious SMA natural history data: A benchmark to measure future treatment successes. Neurology 2018, 91, 337–339. [Google Scholar] [CrossRef]
- Glascock, J.J.; Osman, E.Y.; Wetz, M.J.; Krogman, M.M.; Shababi, M.; Lorson, C.L. Decreasing disease severity in symptomatic, Smn(-/-);SMN2(+/+), spinal muscular atrophy mice following scAAV9-SMN delivery. Hum. Gene Ther. 2012, 23, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Muller-Felber, W.; Vill, K.; Schwartz, O.; Glaser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Roschinger, W.; Becker, M.; Durner, J.; et al. Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening—Opportunity or Burden? J. Neuromuscul. Dis. 2020, 7, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, L.; Solari, A.; Vita, G.; Bertini, E.; Minetti, C.; Mongini, T.; Mazzoni, E.; Angelini, C.; Morandi, L. Role of gabapentin in spinal muscular atrophy: Results of a multicenter, randomized Italian study. J. Child. Neurol. 2003, 18, 537–541. [Google Scholar] [CrossRef]
- Miller, R.G.; Moore, D.H.; Dronsky, V.; Bradley, W.; Barohn, R.; Bryan, W.; Prior, T.W.; Gelinas, D.F.; Iannaccone, S.; Kissel, J.; et al. A placebo-controlled trial of gabapentin in spinal muscular atrophy. J. Neurol. Sci. 2001, 191, 127–131. [Google Scholar] [CrossRef]
- Dimitriadi, M.; Kye, M.J.; Kalloo, G.; Yersak, J.M.; Sahin, M.; Hart, A.C. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci. 2013, 33, 6557–6562. [Google Scholar] [CrossRef] [Green Version]
- Haddad, H.; Cifuentes-Diaz, C.; Miroglio, A.; Roblot, N.; Joshi, V.; Melki, J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 2003, 28, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Abbara, C.; Estournet, B.; Lacomblez, L.; Lelievre, B.; Ouslimani, A.; Lehmann, B.; Viollet, L.; Barois, A.; Diquet, B. Riluzole pharmacokinetics in young patients with spinal muscular atrophy. Br. J. Clin. Pharmacol. 2011, 71, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Pang, K.; Chen, Y.; Wang, S.; Li, H.; Xu, Y.; Han, F.; Yao, H.; Liu, H.; Lopes-Rodrigues, V.; et al. TrkB agonistic antibodies superior to BDNF: Utility in treating motoneuron degeneration. Neurobiol. Dis. 2019, 132, 104590. [Google Scholar] [CrossRef] [PubMed]
- Haase, G.; Kennel, P.; Pettmann, B.; Vigne, E.; Akli, S.; Revah, F.; Schmalbruch, H.; Kahn, A. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat. Med. 1997, 3, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Ikeda, K.; Klinkosz, B.; Cedarbaum, J.M.; Wong, V.; Lindsay, R.M. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 1994, 265, 1107–1110. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Houenou, L.J.; Johnson, J.E.; Lin, L.F.; Li, L.; Lo, A.C.; Newsome, A.L.; Prevette, D.M.; Wang, S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature 1995, 373, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Sendtner, M.; Holtmann, B.; Kolbeck, R.; Thoenen, H.; Barde, Y.A. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 1992, 360, 757–759. [Google Scholar] [CrossRef]
- Sendtner, M.; Schmalbruch, H.; Stockli, K.A.; Carroll, P.; Kreutzberg, G.W.; Thoenen, H. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 1992, 358, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Sendtner, M.; Stockli, K.A.; Thoenen, H. Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J. Cell Biol. 1992, 118, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Murdocca, M.; Malgieri, A.; Luchetti, A.; Saieva, L.; Dobrowolny, G.; de Leonibus, E.; Filareto, A.; Quitadamo, M.C.; Novelli, G.; Musaro, A.; et al. IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol. Med. 2012, 18, 1076–1085. [Google Scholar] [CrossRef]
- Bosch-Marce, M.; Wee, C.D.; Martinez, T.L.; Lipkes, C.E.; Choe, D.W.; Kong, L.; Van Meerbeke, J.P.; Musaro, A.; Sumner, C.J. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum. Mol. Genet. 2011, 20, 1844–1853. [Google Scholar] [CrossRef]
- Shababi, M.; Glascock, J.; Lorson, C.L. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 2011, 22, 135–144. [Google Scholar] [CrossRef]
- Tsai, L.K.; Chen, Y.C.; Cheng, W.C.; Ting, C.H.; Dodge, J.C.; Hwu, W.L.; Cheng, S.H.; Passini, M.A. IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol. Dis. 2012, 45, 272–279. [Google Scholar] [CrossRef]
- Dombert, B.; Balk, S.; Luningschror, P.; Moradi, M.; Sivadasan, R.; Saal-Bauernschubert, L.; Jablonka, S. BDNF/trkB Induction of Calcium Transients through Cav2.2 Calcium Channels in Motoneurons Corresponds to F-actin Assembly and Growth Cone Formation on beta2-Chain Laminin (221). Front. Mol. Neurosci. 2017, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; Balk, S.; Franco-Espin, J.; Ojeda, J.; Hennlein, L.; Drexl, H.; Dombert, B.; Clausen, J.D.; Torres-Benito, L.; Saal-Bauernschubert, L.; et al. R-Roscovitine Improves Motoneuron Function in Mouse Models for Spinal Muscular Atrophy. iScience 2020, 23, 100826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.T.; Chen, C.S.; Jong, Y.J.; Chang, F.R.; Lo, Y.C. Loganin possesses neuroprotective properties, restores SMN protein and activates protein synthesis positive regulator Akt/mTOR in experimental models of spinal muscular atrophy. Pharmacol. Res. 2016, 111, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Yokomaku, D.; Richards, M.; Hori, H.; Adachi, N.; Kunugi, H. Functional interactions between steroid hormones and neurotrophin BDNF. World J. Biol. Chem. 2010, 1, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pinto, A.M.; Wan, L.; Wang, W.; Berg, M.G.; Oliva, I.; Singh, L.N.; Dengler, C.; Wei, Z.; Dreyfuss, G. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 19348–19353. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell. Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Bordet, T.; Berna, P.; Abitbol, J.L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Bertini, E.; Comi, G.; Kirschner, J.; Lusakowska, A.; Mercuri, E.; Scoto, M.; van der Pol, W.L.; Vuillerot, C.; Burdeska, A.; et al. Long-term follow-up of patients with type 2 and non-ambulant type 3 spinal muscular atrophy (SMA) treated with olesoxime in the OLEOS trial. Neuromuscul. Disord. 2020, 30, 959–969. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [Green Version]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Zhu, X.; Hadhazy, M.; Wehling, M.; Tidball, J.G.; McNally, E.M. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 2000, 474, 71–75. [Google Scholar] [CrossRef]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef] [Green Version]
- Rose, F.F., Jr.; Mattis, V.B.; Rindt, H.; Lorson, C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009, 18, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindt, H.; Buckley, D.M.; Vale, S.M.; Krogman, M.; Rose, F.F., Jr.; Garcia, M.L.; Lorson, C.L. Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 2012, 22, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Wee, C.D.; Warsing, L.C.; Choe, D.W.; Ng, A.S.; Lutz, C.; Wagner, K.R. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 2009, 18, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef]
- Dagbay, K.B.; Treece, E.; Streich, F.C., Jr.; Jackson, J.W.; Faucette, R.R.; Nikiforov, A.; Lin, S.C.; Boston, C.J.; Nicholls, S.B.; Capili, A.D.; et al. Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015. J. Biol. Chem. 2020, 295, 5404–5418. [Google Scholar] [CrossRef] [Green Version]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.J.; Hartman, J.J.; Hinken, A.C.; Muci, A.R.; Kawas, R.; Driscoll, L.; Godinez, G.; Lee, K.H.; Marquez, D.; Browne, W.F., IV; et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat. Med. 2012, 18, 452–455. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Edell, S.; Ferrufino, C.; Thomsen, K.; Hwee, D.T.; Morgan, B.P.; Malik, F.I.; Chin, E.R. The Fast Skeletal Muscle Troponin Activator Reldesemtiv in Combination with Nusinersen Improves Muscle Function in a Mouse Model of Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Edell, S.; Ferrufino, C.; Thomsen, K.; Hwee, D.T.; Morgan, B.P.; Malik, F.I.; Chin, E.R. The Fast Skeletal Muscle Troponin Activator Reldesemtiv in Combination with SMN-C1 Improves Muscle Function in a Mouse Model of Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Van der Pol, W.L.; Wadman, R.I.; Van den Berg, L.H.; Vrancken, A.F.J. Dysfunction of the neuromuscular junction in patients with spinal muscular atrophy type 2 and 3. Neuromuscul. Disord. 2012, 22, 871–872. [Google Scholar] [CrossRef]
- Vrbova, G.; Slawinska, U. Critical period of neuromuscular development: Importance for a new treatment of SMA. Neuromuscul. Disord. 2018, 28, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Tarr, T.B.; Malick, W.; Liang, M.; Valdomir, G.; Frasso, M.; Lacomis, D.; Reddel, S.W.; Garcia-Ocano, A.; Wipf, P.; Meriney, S.D. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J. Neurosci. 2013, 33, 10559–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarr, T.B.; Lacomis, D.; Reddel, S.W.; Liang, M.; Valdomir, G.; Frasso, M.; Wipf, P.; Meriney, S.D. Complete reversal of Lambert-Eaton myasthenic syndrome synaptic impairment by the combined use of a K+ channel blocker and a Ca2+ channel agonist. J. Physiol. 2014, 592, 3687–3696. [Google Scholar] [CrossRef] [PubMed]
- Ojala, K.S.; Liang, M.; Wipf, P.; Meriney, S.D. A Novel, Peripherally-Targeted Treatment for Ameliorating the Neuromuscular Weakness Caused by Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Ojala, K.S.; Kaufhold, C.J.; Davey, M.R.; Liang, M.; Wipf, P.; Meriney, S.D. GV-58, a selective calcium channel gating modifier, can increase muscle strength and neurotransmission in untreated and ASO-treated SMA model mice. In Proceedings of the Cure SMA Conference, Orlando, FL, USA, 11–14 June 2020. [Google Scholar]
- Bernal, S.; Alias, L.; Barcelo, M.J.; Also-Rallo, E.; Martinez-Hernandez, R.; Gamez, J.; Guillen-Navarro, E.; Rosell, J.; Hernando, I.; Rodriguez-Alvarez, F.J.; et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 2010, 47, 640–642. [Google Scholar] [CrossRef] [Green Version]
- Bernal, S.; Also-Rallo, E.; Martinez-Hernandez, R.; Alias, L.; Rodriguez-Alvarez, F.J.; Millan, J.M.; Hernandez-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef]
- Burghes, A.H.; Ingraham, S.E.; Kote-Jarai, Z.; Rosenfeld, S.; Herta, N.; Nadkarni, N.; DiDonato, C.J.; Carpten, J.; Hurko, O.; Florence, J.; et al. Linkage mapping of the spinal muscular atrophy gene. Hum. Genet. 1994, 93, 305–312. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alias, L.; March, F.; Vencesla, A.; Rodriguez-Alvarez, F.J.; Aller, E.; Fernandez, R.M.; Borrego, S.; Millan, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Cobben, J.M.; van der Steege, G.; Grootscholten, P.; de Visser, M.; Scheffer, H.; Buys, C.H. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 1995, 57, 805–808. [Google Scholar] [PubMed]
- DiDonato, C.J.; Morgan, K.; Carpten, J.D.; Fuerst, P.; Ingraham, S.E.; Prescott, G.; McPherson, J.D.; Wirth, B.; Zerres, K.; Hurko, O.; et al. Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am. J. Hum. Genet. 1994, 55, 1218–1229. [Google Scholar]
- Hahnen, E.; Forkert, R.; Marke, C.; Rudnik-Schoneborn, S.; Schonling, J.; Zerres, K.; Wirth, B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 1995, 4, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejowska, M.; Borkowska, J.; Zimowski, J.; Kostera-Pruszczyk, A.; Milewski, M.; Jurek, M.; Sielska, D.; Kostyk, E.; Nyka, W.; Zaremba, J.; et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur. J. Hum. Genet. 2008, 16, 930–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedrzejowska, M.; Milewski, M.; Zimowski, J.; Borkowska, J.; Kostera-Pruszczyk, A.; Sielska, D.; Jurek, M.; Hausmanowa-Petrusewicz, I. Phenotype modifiers of spinal muscular atrophy: The number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease. Acta Biochim. Pol. 2009, 56, 103–108. [Google Scholar] [CrossRef]
- Oprea, G.E.; Krober, S.; McWhorter, M.L.; Rossoll, W.; Muller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Pane, M.; Lapenta, L.; Abiusi, E.; de Sanctis, R.; Luigetti, M.; Palermo, C.; Ranalli, D.; Fiori, S.; Tiziano, F.D.; Mercuri, E. Longitudinal assessments in discordant twins with SMA. Neuromuscul. Disord. 2017, 27, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Prior, T.W.; Swoboda, K.J.; Scott, H.D.; Hejmanowski, A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A 2004, 130A, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frebourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadman, R.I.; Jansen, M.D.; Curial, C.A.D.; Groen, E.J.N.; Stam, M.; Wijngaarde, C.A.; Medic, J.; Sodaar, P.; van Eijk, K.R.; Huibers, M.M.H.; et al. Analysis of FUS, PFN2, TDP-43, and PLS3 as potential disease severity modifiers in spinal muscular atrophy. Neurol. Genet. 2020, 6, e386. [Google Scholar] [CrossRef] [Green Version]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Lohr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef] [Green Version]
- Torres-Benito, L.; Schneider, S.; Rombo, R.; Ling, K.K.; Grysko, V.; Upadhyay, A.; Kononenko, N.L.; Rigo, F.; Bennett, C.F.; Wirth, B. NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice. Am. J. Hum. Genet. 2019, 105, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrafiah, A.; Karyka, E.; Coldicott, I.; Iremonger, K.; Lewis, K.E.; Ning, K.; Azzouz, M. Plastin 3 Promotes Motor Neuron Axonal Growth and Extends Survival in a Mouse Model of Spinal Muscular Atrophy. Mol. Ther. Methods Clin. Dev. 2018, 9, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Delanote, V.; Vandekerckhove, J.; Gettemans, J. Plastins: Versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 2005, 26, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engqvist-Goldstein, A.E.; Drubin, D.G. Actin assembly and endocytosis: From yeast to mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 287–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Kaifer, K.A.; Villalon, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.T.; Wolman, M.; Granato, M.; Beattie, C.E. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J. Neurosci. 2012, 32, 5074–5084. [Google Scholar] [CrossRef] [Green Version]
- McGovern, V.L.; Massoni-Laporte, A.; Wang, X.; Le, T.T.; Le, H.T.; Beattie, C.E.; Rich, M.M.; Burghes, A.H. Plastin 3 Expression Does Not Modify Spinal Muscular Atrophy Severity in the 7 SMA Mouse. PLoS ONE 2015, 10, e0132364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.Y.; Mikhail, A.; Ljubicic, V. Mechanisms of exercise-induced survival motor neuron expression in the skeletal muscle of spinal muscular atrophy-like mice. J. Physiol. 2019, 597, 4757–4778. [Google Scholar] [CrossRef]
- Grondard, C.; Biondi, O.; Armand, A.S.; Lecolle, S.; Della Gaspera, B.; Pariset, C.; Li, H.; Gallien, C.L.; Vidal, P.P.; Chanoine, C.; et al. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2005, 25, 7615–7622. [Google Scholar] [CrossRef] [Green Version]
- Biondi, O.; Grondard, C.; Lecolle, S.; Deforges, S.; Pariset, C.; Lopes, P.; Cifuentes-Diaz, C.; Li, H.; della Gaspera, B.; Chanoine, C.; et al. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2008, 28, 953–962. [Google Scholar] [CrossRef]
- Charbonnier, F. Exercise-induced neuroprotection in SMA model mice: A means for determining new therapeutic strategies. Mol. Neurobiol. 2007, 35, 217–223. [Google Scholar] [CrossRef]
- Bora, G.; Subasi-Yildiz, S.; Yesbek-Kaymaz, A.; Bulut, N.; Alemdaroglu, I.; Tunca-Yilmaz, O.; Topaloglu, H.; Karaduman, A.A.; Erdem-Yurter, H. Effects of Arm Cycling Exercise in Spinal Muscular Atrophy Type II Patients: A Pilot Study. J. Child Neurol. 2018, 33, 209–215. [Google Scholar] [CrossRef]
- Houdebine, L.; D’Amico, D.; Bastin, J.; Chali, F.; Desseille, C.; Rumeau, V.; Soukkari, J.; Oudot, C.; Rouquet, T.; Bariohay, B.; et al. Low-Intensity Running and High-Intensity Swimming Exercises Differentially Improve Energy Metabolism in Mice With Mild Spinal Muscular Atrophy. Front. Physiol. 2019, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Chali, F.; Desseille, C.; Houdebine, L.; Benoit, E.; Rouquet, T.; Bariohay, B.; Lopes, P.; Branchu, J.; Della Gaspera, B.; Pariset, C.; et al. Long-term exercise-specific neuroprotection in spinal muscular atrophy-like mice. J. Physiol. 2016, 594, 1931–1952. [Google Scholar] [CrossRef] [Green Version]
- Lewelt, A.; Krosschell, K.J.; Stoddard, G.J.; Weng, C.; Xue, M.; Marcus, R.L.; Gappmaier, E.; Viollet, L.; Johnson, B.A.; White, A.T.; et al. Resistance strength training exercise in children with spinal muscular atrophy. Muscle Nerve 2015, 52, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, K.L.; Hansen, R.S.; Preisler, N.; Thogersen, F.; Berthelsen, M.P.; Vissing, J. Training improves oxidative capacity, but not function, in spinal muscular atrophy type III. Muscle Nerve 2015, 52, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Bartels, B.; Montes, J.; van der Pol, W.L.; de Groot, J.F. Physical exercise training for type 3 spinal muscular atrophy. Cochrane Database Syst. Rev. 2019, 3, CD012120. [Google Scholar] [CrossRef] [Green Version]
- Vita, G.L.; Stancanelli, C.; La Foresta, S.; Faraone, C.; Sframeli, M.; Ferrero, A.; Fattore, C.; Galbo, R.; Ferraro, M.; Ricci, G.; et al. Psychosocial impact of sport activity in neuromuscular disorders. Neurol. Sci. 2020, 41, 2561–2567. [Google Scholar] [CrossRef]
- Gandolla, M.; Antonietti, A.; Longatelli, V.; Pedrocchi, A. The Effectiveness of Wearable Upper Limb Assistive Devices in Degenerative Neuromuscular Diseases: A Systematic Review and Meta-Analysis. Front. Bioeng. Biotechnol. 2019, 7, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagengruber, A.; Vogel, J. Functional Tasks Performed by People with Severe Muscular Atrophy Using an sEMG Controlled Robotic Manipulator. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2018, 2018, 1713–1718. [Google Scholar] [CrossRef]
- Haumont, T.; Rahman, T.; Sample, W.; King, M.M.; Church, C.; Henley, J.; Jayakumar, S. Wilmington robotic exoskeleton: A novel device to maintain arm improvement in muscular disease. J. Pediatr. Orthop. 2011, 31, e44–e49. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Sample, W.; Seliktar, R.; Alexander, M.; Scavina, M. A body-powered functional upper limb orthosis. J. Rehabil. Res. Dev. 2000, 37, 675–680. [Google Scholar]
- Sanz-Merodio, D.; Puyuelo, G.; Ganguly, A.; Garces, E.; Goñi, A.; Garcia, E. EXOtrainer Project Clinical Evaluation of Gait Training with Exoskeleton in Children with Spinal Muscular Atrophy. In Advances in Robotics Research: From Lab to Market: ECHORD++: Robotic Science Supporting Innovation; Grau, A., Morel, Y., Puig-Pey, A., Cecchi, F., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 211–227. [Google Scholar] [CrossRef]
- Sakaki, T.; Aoki, K.; Ushijima, K.; Sakuragi, M. Robotic stretcher for spinal muscular atrophy patient: Preliminary tests of user controllability. In Proceedings of the 10th International Conference on Ubiquitous Robots and Ambient Intelligence (URAI), Jeju, Korea, 30 October–2 November 2013; pp. 156–160. [Google Scholar]
- Pera, M.C.; Luigetti, M.; Pane, M.; Coratti, G.; Forcina, N.; Fanelli, L.; Mazzone, E.S.; Antonaci, L.; Lapenta, L.; Palermo, C.; et al. 6MWT can identify type 3 SMA patients with neuromuscular junction dysfunction. Neuromuscul. Disord. 2017, 27, 879–882. [Google Scholar] [CrossRef]
- Wadman, R.I.; Vrancken, A.F.; van den Berg, L.H.; van der Pol, W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.M.; Montes, J.; Finkel, R.S. Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological Exam-Part 2: Experience from a nusinersen clinical study. Muscle Nerve 2018, 57, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.M. Nusinersen: Antisense oligonucleotide to increase SMN protein production in spinal muscular atrophy. Drugs Today (Barc) 2017, 53, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.D.; Severyn, S.; Linsenmayer, M.; Kelly, K.; Bartlett, A.; Tellez, M.; Heintzman, S.; Sterling, G.; Kolb, S.J.; Elsheikh, B. NMJ Transmission Failure in Adults with Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Orlando, FL, USA, 11–14 June 2020. [Google Scholar]
- Je, H.S.; Yang, F.; Ji, Y.; Potluri, S.; Fu, X.Q.; Luo, Z.G.; Nagappan, G.; Chan, J.P.; Hempstead, B.; Son, Y.J.; et al. ProBDNF and mature BDNF as punishment and reward signals for synapse elimination at mouse neuromuscular junctions. J. Neurosci. 2013, 33, 9957–9962. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Differential pre-mRNA splicing of SMN1 and SMN2 genes. The SMN1 gene (right) effectively splices exon 7 into the mature mRNA transcript, resulting in ~100% full-length SMN protein (blue full circles). In contrast, a C to T nucleotide transition in exon 7 of the SMN2 gene (left; resulting in a U nucleotide in exon 7 pre-mRNA) causes ~90% of the mature mRNA transcripts to lack exon 7 (pink incomplete circles). Without exon 7, the truncated protein (SMNΔ7) is unstable, ineffective at oligomerization, and consequently degraded.
Figure 1.
Differential pre-mRNA splicing of SMN1 and SMN2 genes. The SMN1 gene (right) effectively splices exon 7 into the mature mRNA transcript, resulting in ~100% full-length SMN protein (blue full circles). In contrast, a C to T nucleotide transition in exon 7 of the SMN2 gene (left; resulting in a U nucleotide in exon 7 pre-mRNA) causes ~90% of the mature mRNA transcripts to lack exon 7 (pink incomplete circles). Without exon 7, the truncated protein (SMNΔ7) is unstable, ineffective at oligomerization, and consequently degraded.

Figure 2.
General cellular functions of SMN in motoneurons. SMN has several proposed functions in the motoneuron, though which roles are associated with which specific pathologies are unclear. SMN functions in RNA metabolism, protein homeostasis, and cytoskeleton dynamics. SMN plays additional roles in axonal transport of mRNA and synaptic vesicles, axiogenesis, and axon pathfinding. At the neuromuscular synapse, SMN is involved in actin dynamics, vesicle release and recycling, and neuromuscular junction (NMJ) maturation and repair. These cellular functions of SMN have been thoroughly discussed elsewhere [52,56,70,71,72,73,74].
Figure 2.
General cellular functions of SMN in motoneurons. SMN has several proposed functions in the motoneuron, though which roles are associated with which specific pathologies are unclear. SMN functions in RNA metabolism, protein homeostasis, and cytoskeleton dynamics. SMN plays additional roles in axonal transport of mRNA and synaptic vesicles, axiogenesis, and axon pathfinding. At the neuromuscular synapse, SMN is involved in actin dynamics, vesicle release and recycling, and neuromuscular junction (NMJ) maturation and repair. These cellular functions of SMN have been thoroughly discussed elsewhere [52,56,70,71,72,73,74].

Figure 3.
Developmental regulation and requirement of SMN expression in mice. SMN expression is highest during the perinatal period (indicated by an orange to white gradient), but precipitously drops after development of neuromuscular junctions (NMJs; at approximately 17–20 days of age). After the critical period of neuromuscular development, NMJs are viable with low SMN levels. In adulthood, remodeling after injury requires elevated SMN levels to recover and stabilize NMJs. Current therapies for SMA are restricted to the postnatal period. Schematic adapted with permission from Kariya et al., 2014 [85].
Figure 3.
Developmental regulation and requirement of SMN expression in mice. SMN expression is highest during the perinatal period (indicated by an orange to white gradient), but precipitously drops after development of neuromuscular junctions (NMJs; at approximately 17–20 days of age). After the critical period of neuromuscular development, NMJs are viable with low SMN levels. In adulthood, remodeling after injury requires elevated SMN levels to recover and stabilize NMJs. Current therapies for SMA are restricted to the postnatal period. Schematic adapted with permission from Kariya et al., 2014 [85].

Figure 4.
FDA-approved SMN-based therapies for SMA. (A) In the absence of therapeutic intervention, the SMN2 gene produces predominantly truncated SMN protein (pink incomplete circle) and only a small percentage of full-length SMN protein (blue full circle) due to poor inclusion of exon 7 in mature mRNA transcripts. (B) The first FDA-approved therapy for SMA utilizes antisense oligonucleotides (ASOs; green icon; brand name Spinraza®) to target intronic splicing silencer N1 (ISS-N1), thereby increasing inclusion of exon 7 in mature mRNA transcripts. (C) The most recent FDA-approved therapy, risdiplam (purple circle; brand name Evrysdi™), enhances U1-pre-mRNA association to promote inclusion of exon 7 in mature mRNA transcripts. (D) SMA is caused by homozygous loss of functional SMN1 (by gene deletion or conversion), resulting in a lack of SMN protein derived from this gene. (E) Onasemnogene-xioi (brand name Zolgensma®) causes transduced cells to transcribe full-length SMN cDNA.
Figure 4.
FDA-approved SMN-based therapies for SMA. (A) In the absence of therapeutic intervention, the SMN2 gene produces predominantly truncated SMN protein (pink incomplete circle) and only a small percentage of full-length SMN protein (blue full circle) due to poor inclusion of exon 7 in mature mRNA transcripts. (B) The first FDA-approved therapy for SMA utilizes antisense oligonucleotides (ASOs; green icon; brand name Spinraza®) to target intronic splicing silencer N1 (ISS-N1), thereby increasing inclusion of exon 7 in mature mRNA transcripts. (C) The most recent FDA-approved therapy, risdiplam (purple circle; brand name Evrysdi™), enhances U1-pre-mRNA association to promote inclusion of exon 7 in mature mRNA transcripts. (D) SMA is caused by homozygous loss of functional SMN1 (by gene deletion or conversion), resulting in a lack of SMN protein derived from this gene. (E) Onasemnogene-xioi (brand name Zolgensma®) causes transduced cells to transcribe full-length SMN cDNA.


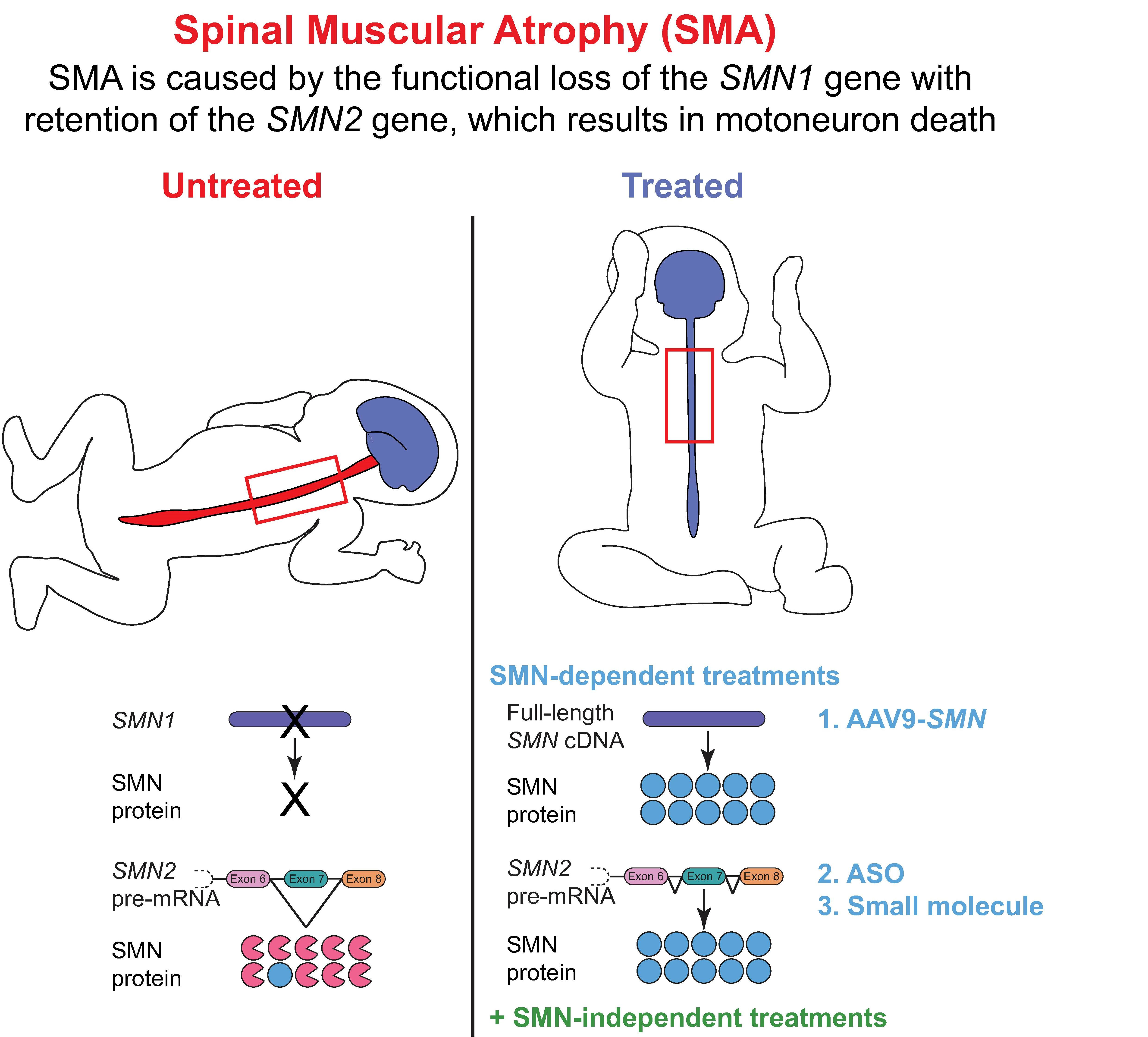{kind=link}
{kind=link}
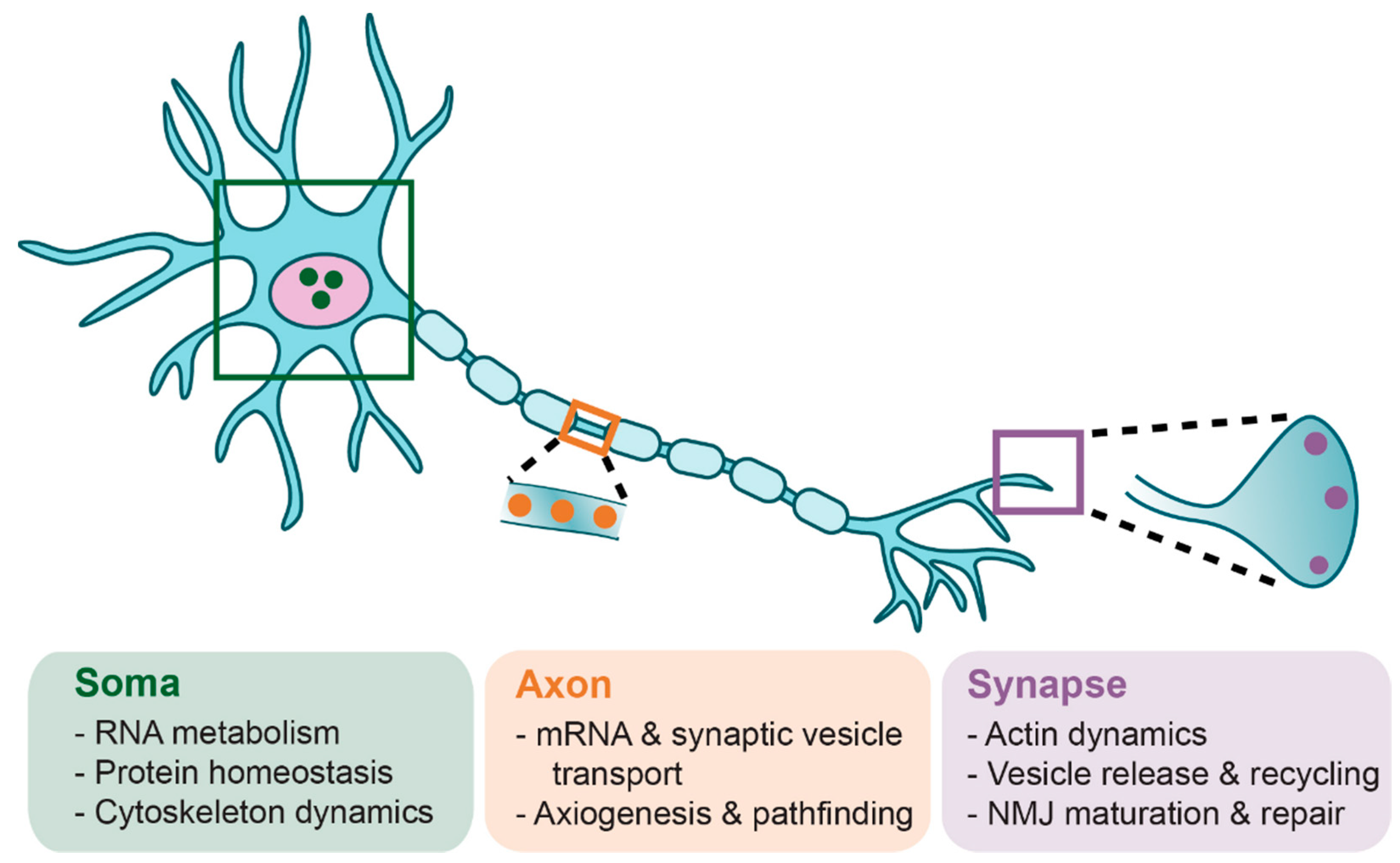{kind=link}
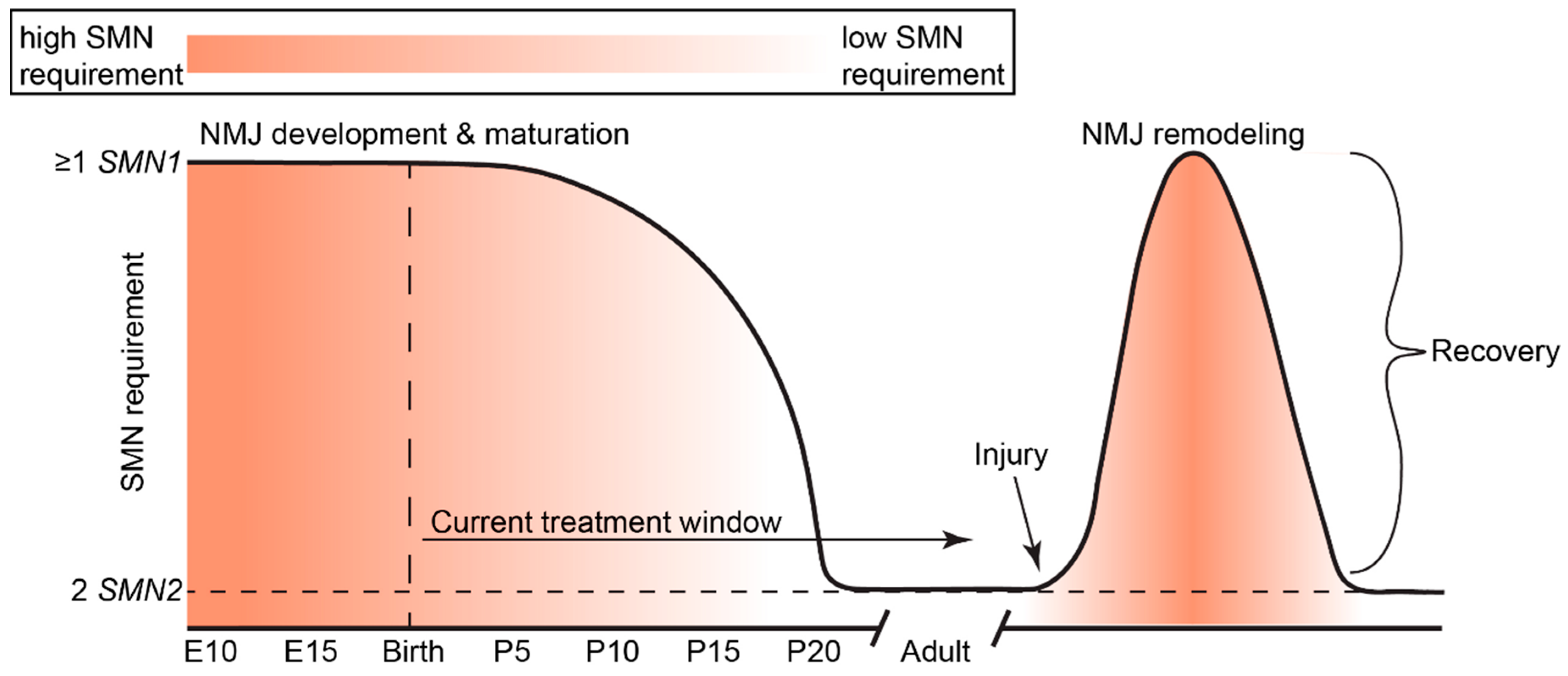{kind=link}
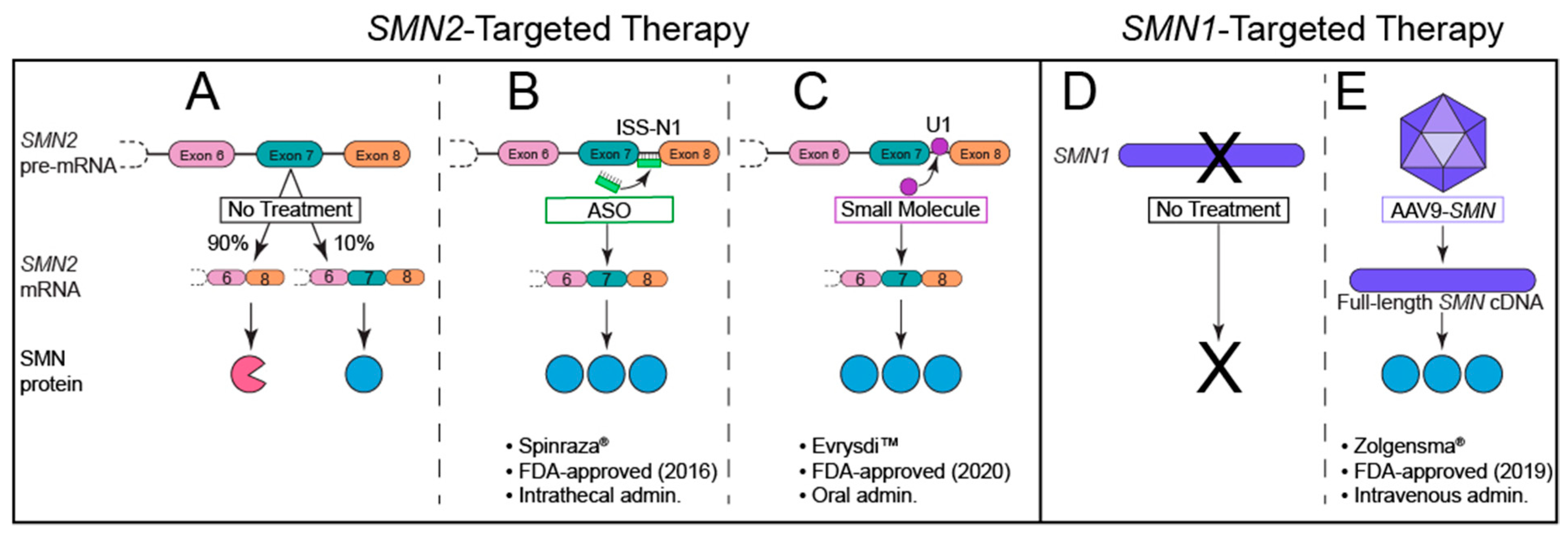{kind=link}
Table 1.
Natural histories of Type 0–Type IV SMA.
| SMA Type | Age of Symptom Onset | Defining Motor Function | Common Characteristics | Life Expectancy | SMN2 Copy Number |
|---|---|---|---|---|---|
| Type 0 | Prenatal | Respiratory support |
| ˂6 months | 1 |
| Type I | 0–6 months | Never sits unsupported |
| ˂2 years | 2 |
| Type II | 6–18 months | Sits; never stands independently |
| >2 years | 2–3 |
| Type III | ˃18 months | Walks |
| Adult | 3–4 |
| Type IV | ˃21 years | All motor function |
| Adult | ≥4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ojala, K.S.; Reedich, E.J.; DiDonato, C.J.; Meriney, S.D. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sci. 2021, 11, 194. https://doi.org/10.3390/brainsci11020194
AMA Style
Ojala KS, Reedich EJ, DiDonato CJ, Meriney SD. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sciences. 2021; 11(2):194. https://doi.org/10.3390/brainsci11020194
Chicago/Turabian StyleOjala, Kristine S., Emily J. Reedich, Christine J. DiDonato, and Stephen D. Meriney. 2021. "In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy" Brain Sciences 11, no. 2: 194. https://doi.org/10.3390/brainsci11020194
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.